NR 602 MIDTERM EXAMS
STUDY GUIDE
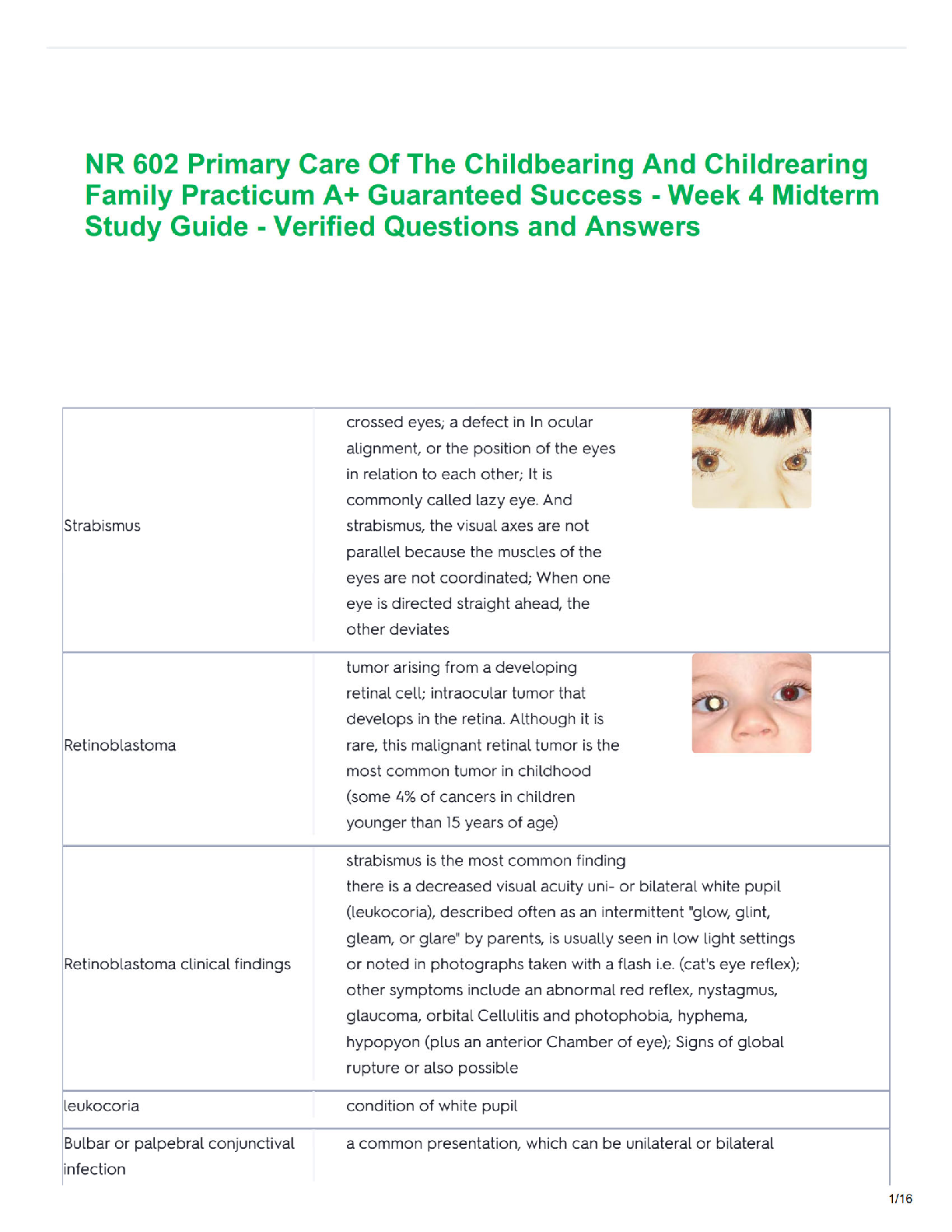
Chalazion
Chalazion is a chronic sterile inflammation of the eyelid resulting from a lipogranuloma of the meibomian glands that line the
posterior margins of the eyelids (see Fig. 29-
...
NR 602 MIDTERM EXAMS
STUDY GUIDE
Chalazion
Chalazion is a chronic sterile inflammation of the eyelid resulting from a lipogranuloma of the meibomian glands that line the
posterior margins of the eyelids (see Fig. 29-7). It is deeper in the eyelid tissue than a hordeolum and may result from an internal
hordeolum or retained lipid granular secretions.
Clinical Findings
Initially, mild erythema and slight swelling of the involved eyelid are seen. After a few days the inflammation resolves, and a slow growing,
round, nonpigmented, painless (key finding) mass remains. It may persist for a long time and is a commonly acquired lid lesion seen in
children (see Fig. 29-7).
727
Management
• Acute lesions are treated with hot compresses.
• Refer to an ophthalmologist for surgical incision or topical intralesional corticosteroid injections if the condition is unresolved or if the
lesion causes cosmetic concerns. A chalazion can distort vision by causing astigmatism as a result of pressure on the orbit.
Complications
Recurrence is common. Fragile, vascular granulation tissue called pyogenic granuloma that enlarges and bleeds rapidly can occur if a
chalazion breaks through the conjunctival surface.
Blepharitis
Blepharitis is an acute or chronic inflammation of the eyelash follicles or meibomian sebaceous glands of the eyelids (or both). It is usually
bilateral. There may be a history of contact lens wear or physical contact with another symptomatic person. It is commonly caused by
contaminated makeup or contact lens solution. Poor hygiene, tear deficiency, rosacea, and seborrheic dermatitis of the scalp and face are also
possible etiologic factors. The ulcerative form of blepharitis is usually caused by S. aureus. Nonulcerative blepharitis is occasionally seen in
children with psoriasis, seborrhea, eczema, allergies, lice infestation, or in children with trisomy 21.
Clinical Findings
• Swelling and erythema of the eyelid margins and palpebral conjunctiva
726
• Flaky, scaly debris over eyelid margins on awakening; presence of lice
• Gritty, burning feeling in eyes
• Mild bulbar conjunctival injection
• Ulcerative form: Hard scales at the base of the lashes (if the crust is removed, ulceration is seen at the hair follicles, the lashes fall out, and
an associated conjunctivitis is present)
Differential Diagnosis
Pediculosis of the eyelashes.
Management
Explain to the patient that this may be chronic or relapsing. Instructions for the patient include:
• Scrub the eyelashes and eyelids with a cotton-tipped applicator containing a weak (50%) solution of no-tears shampoo to maintain proper
hygiene and debride the scales.
• Use warm compresses for 5 to 10 minutes at a time two to four times a day and wipe away lid debris.
• At times antistaphylococcal antibiotic (e.g., erythromycin 0.5% ophthalmic ointment) is used until symptoms subside and for at least 1 week
thereafter. Ointment is preferable to eye drops because of increased duration of contact with the ocular tissue. Azithromycin 1% ophthalmic
solution for 4 weeks may also be used (Shtein, 2014).
• Treat associated seborrhea, psoriasis, eczema, or allergies as indicated.
• Remove contact lenses and wear eyeglasses for the duration of the treatment period. Sterilize or clean lenses before reinserting.• Purchase new eye makeup; minimize use of mascara and eyeliner.
• Use artificial tears for patients with inadequate tear pools.
Chronic staphylococcal blepharitis and meibomian keratoconjunctivitis respond to oral erythromycin. Doxycycline, tetracycline, or
minocycline can be used chronically in children older than 8 years old.
Acute Otitis Media
AOM is an acute infection of the middle ear (Fig. 30-4). The AAP Clinical Practice Guideline requires the presence of the
following three components to diagnose AOM (Lieberthal et al, 2013):
• Recent, abrupt onset of signs and symptoms of middle ear inflammation and effusion (ear pain, irritability, otorrhea, and/or
fever)
• MEE as confirmed by bulging TM, limited or absent mobility by pneumatic otoscopy, air-fluid level behind TM, and/or
otorrhea
• Signs and symptoms of middle ear inflammation as confirmed by distinct erythema of the TM or onset of ear pain (holding,
tugging, rubbing of the ear in a nonverbal manner)
Characteristics of different types of AOM are defined in Table 30-4. AOM often follows eustachian tube dysfunction (ETD).
Common causes of ETD include upper respiratory infections, allergies, and ETS. ETD leads to 746functional eustachian tube
obstruction and inflammation that decreases the protective ciliary action in the eustachian tube. When the eustachian tube is
obstructed, negative pressure develops as air is absorbed in the middle ear (see Fig. 30-4). The negative pressure pulls fluid from
the mucosal lining and causes an accumulation of sterile fluid. Bacteria pulled in from the eustachian tube lead to the
accumulation of purulent fluid. Young children have shorter, more horizontal and more flaccid eustachian tubes that are easily
disrupted by viruses, which predisposes them to AOM. Respiratory syncytial virus and influenza are two of the viruses most
responsible for the increase in the incidence of AOM seen from January to April. Other risk factors associated with AOM are
listed in Boxes 30-1 and 30-2.
S. pneumoniae, nontypeable Haemophilus influenzae, Moraxella catarrhalis, and S. pyogenes (group A streptococci) are the most
common infecting organisms in AOM (Conover, 2013). S. pneumoniaecontinues to be the most common bacteria responsible for
AOM. The strains of S. pneumoniae in the heptavalent pneumococcal conjugate vaccine (PCV7) have virtually disappeared from
the middle ear fluid of children with AOM (Lieberthal et al, 2013). With the introduction of the 13-valent S. pneumoniae vaccine,
the bacteriology of the middle ear is likely to continue to evolve. Bullous myringitis is almost always caused by S. pneumonia.
Nontypeable H. influenza remains a common cause of AOM. It is the most common cause of bilateral otitis media, severe
inflammation of the TM, and otitis-conjunctivitis syndrome. M. catarrhalis obtained from the nasopharynx has become
increasingly more beta-lactamase positive, but the high rate of clinical resolution in children with AOM from M.
catarrhalis makes amoxicillin a good choice for initial therapy (Lieberthal et al, 2013). M. catarrhalis rarely causes invasive
disease. S. pyogenes is responsible for AOM in older children, is responsible for more TM ruptures, and is more likely to cause
mastoiditis.
Although a virus is usually the initial causative factor in AOM, strict diagnostic criteria, careful specimen handling, and
sensitive microbiologic techniques have shown that the majority of AOM is caused by bacteria or bacteria and virus together
(Lieberthal et al, 2013).
Clinical Findings
History
Rapid onset of signs and symptoms:
• Ear pain with possible ear pulling in the infant; may interfere with activity and/or sleep
• Irritability in an infant or toddler
• Otorrhea
• Fever
Other key factors or symptoms:
• Prematurity
• Craniofacial anomalies or congenital syndromes associated with craniofacial anomalies
• Exposure to risk factors
• Disrupted sleep or inability to sleep
• Lethargy, dizziness, tinnitus, and unsteady gait• Diarrhea and vomiting
• Sudden hearing loss
• Stuffy nose, rhinorrhea, and sneezing
• Rare facial palsy and ataxia
Physical Examination
• Presence of MEE, confirmed by pneumatic otoscopy, tympanometry, or acoustic reflectometry, as evidenced by:
• Bulging TM (see Fig. 30-4)
• Decreased translucency of TM
• Absent or decreased mobility of the TM
• Air-fluid level behind the TM
• Otorrhea
747
• Signs and symptoms of middle ear inflammation indicated by either:
• Erythema of the TM (Amber is usually seen in otitis media with effusion [OME]; white or yellow may be seen in either AOM or OME
[Shaikh et al, 2010].)
or
• Distinct otalgia that interferes with normal activity or sleep
• In addition, the following TM findings may be present:
• Increased vascularity with obscured or absent landmarks (see Fig. 30-4).
• Red, yellow, or purple TM (Redness alone should not be used to diagnose AOM, especially in a crying child.)
• Thin-walled, sagging bullae filled with straw-colored fluid seen with bullous myringitis
Diagnostic Studies
Pneumatic otoscopy is the simplest and most efficient way to diagnose AOM. Tympanometry reflects effusion (type B pattern).
Tympanocentesis to identify the infecting organism is helpful in the treatment of infants younger than 2 months old. In older infants and
children, tympanocentesis is rarely done and is useful only if the patient is toxic or immunocompromised or in the presence of resistant
infection or acute pain from bullous myringitis. If a tympanocentesis is warranted, refer the patient to an otolaryngologist for this procedure.
Differential Diagnosis
OME, mastoiditis, dental abscess, sinusitis, lymphadenitis, parotitis, peritonsillar abscess, trauma, ETD, impacted teeth, temporomandibular
joint dysfunction, and immune deficiency are differential diagnoses. Any infant 2 months old or younger with AOM should be evaluated for
fever without focus and not just treated for an ear infection.
Management
Many changes have been made in the treatment of AOM because of the increasing rate of antibiotic-resistant bacteria related to the
injudicious use of antibiotics. Ample evidence has been presented that symptom management may be all that is required in children with
MEE without other symptoms of AOM (Lieberthal et al, 2013). Treatment guidelines are decided based on the child's age, illness severity,
and the certainty of diagnosis. Table 30-5 shows the recommendation for the diagnosis and subsequent treatment of AOM.
1. Pain management is the first principle of treatment.
• Weight-appropriate doses of ibuprofen or acetaminophen should be encouraged to decrease discomfort and fever.
• Topical analgesics, such as benzocaine or antipyrine/benzocaine otic preparations, can be added to systemic pain management if the
TM is known to be intact. Topical analgesics should not be used alone.
• Distraction, oil application, or external use of heat or cold may be of some use.
2. Antibiotics are also effective. (Table 30-6 lists dosage recommendations.)
• Amoxicillin remains the first-line antibiotic for AOM if there has not been a previous treated AOM in the previous 30 days, there is
no conjunctivitis, and no penicillin allergy (Lieberthal et al, 2013). Beta-lactam coverage (amoxicillin/clavulanate, thirdgeneration cephalosporin) is recommended when the child has been treated with amoxicillin in the previous 30 days, there is an
allergy to penicillin, and the child has concurrent conjunctivitis or has recurrent otitis that has not responded to amoxicillin. If
there is a documented hypersensitivity reaction to amoxicillin, the following antibiotics are acceptable, follow the non-type 1
hypersensitivity and type 1 hypersensitivity recommendations in Table 30-6:
• Ceftriaxone may be effective for the vomiting child, the child unable to tolerate oral medications, or the child who has failed
amoxicillin/clavulanate.
748
• Clindamycin may be considered for ceftriaxone failure but should only be used if susceptibilities are known.
• Prophylactic antibiotics for chronic or recurrent AOM are not recommended.
3. Observation or ―watchful waiting‖ for 48 to 72 hours (see Table 30-5) allows the patient to improve without antibiotic treatment. Pain
relief should be provided, and a means of follow-up must be in place. Options for follow-up include:
• Parent-initiated visit or phone call for worsening or no improvement
• Scheduled follow-up appointment
• Routine follow-up phone call• Given a prescription to be started if the child's symptoms do not improve or if they worsen in 48 to 72 hours (Table 30-7)
• Communication with the parent, reevaluation, and the ability to obtain medication must be in place.
4. Recommendations for follow-up include:
• After 48 to 72 hours if a child has not showed improvement in ear symptomatology, the child should be seen to confirm or exclude
the presence of AOM. If the initial management option was an antibacterial agent, the agent should be changed.
Prevention and Education
The following interventions, shown to be helpful in preventing AOM, should be encouraged:
• Exclusive breastfeeding until at least 6 months of age seems to be protective against AOM
• Avoid bottle propping, feeding infants lying down, and passive smoke exposure
• Avoid the use of pacifiers: Although the relationship cannot be fully explained, multiple studies have shown that pacifier use
increases the incidence of AOM (Lieberthal et al, 2013).
• Pneumococcal vaccine; specifically PCV13, which contains subtype 19A
• Annual influenza vaccine may help prevent otitis media
750
• Xylitol liquid or chewing gum as tolerated
• Choose licensed day care facilities with fewer children
• Educate regarding the problem of drug-resistant bacteria and the need to avoid the use of antibiotics unless absolutely
necessary; if antibiotics are used, the child needs to complete the entire course of the prescription and follow up if symptoms
do not resolve
Conjunctivitis
An estimated 6 million cases of bacterial conjunctivitis occur in the United States annually, at an estimated cost of $377 million
to $857 million (Azari and Barney, 2013). Conjunctivitis is an inflammation of the palpebral and occasionally the bulbar
conjunctiva (Fig. 29-5). It is the most frequently seen ocular disorder in pediatric practice. In pediatric patients, bacteria are the
most common cause of infection (50% to 75%) most commonly from December to April. Pathogens include H. influenzae,
Streptococcus pneumoniae, and Moraxella species with both gram-negative and gram-positive organisms implicated (Azari and
Barney, 2013).
Conjunctivitis also occurs as a viral or fungal infection or as a response to allergens or chemical irritants. Bacterial
conjunctivitis is often unilateral, whereas viral conjunctivitis is most often bilateral. Unilateral disease can also suggest a toxic,
chemical, mechanical, or lacrimal cause. Blockage of the tear drainage system (e.g., from meibomianitis or blepharitis), injury,
foreign body, abrasion or ulcers, keratitis, iritis, herpes simplex virus (HSV), and infantile glaucoma are other known causes.
Patient age is a major indicator of etiology (Table 29-6).
Types of Conjunctivitis
Type Incidence/Etiology Clinical Findings Diagnosis Management*
Ophthalmia
neonatorum
Neonates: Chlamydia trachomatis,
Staphylococcus aureus, Neisseria
gonorrhoeae, HSV (silver nitrate
reaction occurs in 10% of neonates)
Erythema, chemosis,
purulent
exudate
with N.
gonorrhoeae;
clear to mucoid
exudate with
chlamydia
Culture (ELISA, PCR), Gram
stain, R/O N.
gonorrhoeae,chlamydia
Saline irrigation to eyes until
exudate gone; follow with
erythromycin ointment
For N.
gonorrhoeae:ceftriaxone
or IM or IV
For chlamydia: erythromycin
or possibly azithromycin
PO
For HSV: antivirals IV or PO
Bacterial
conjunctivitis
In neonates 5 to 14 days old, preschoolers,
and sexually active teens: Haemophilus
influenzae(nontypeable), Streptococcus
pneumoniae, S. aureus, N. gonorrhoeae
Erythema, chemosis,
itching,
burning,
mucopurulent
exudate, matter
in eyelashes; ↑
Cultures (required in neonate);
Gram stain (optional);
chocolate agar (for N.
gonorrhoeae) R/O
pharyngitis, N.
gonorrhoeae, AOM,
Neonates: Erythromycin
0.5% ophthalmic ointment
≥1 year old: Fourthgeneration fluoroquinolone
For concurrent AOM: Treat
accordingly for AOM
Warm soaks to eyes three
times a day until clearType Incidence/Etiology Clinical Findings Diagnosis Management*
in winter URI, seborrhea No sharing towels, pillows
No school until treatment
begins
Chronic bacterial
conjunctivitis
(unresponsiv
e
conjunctivitis
previously
treated as
bacterial in
etiology)
School-age children and teens: Bacteria,
viruses, C. trachomatis
Same as above;
foreign body
sensation
Cultures, Gram stain; R/O
dacryostenosis,
blepharitis, corneal
ulcers, trachoma
Depends on prior treatment,
laboratory results, and
differential diagnoses
Review compliance and prior
drug choices of
conjunctivitis treatment
Consult with ophthalmologist
Inclusion
conjunctivitis
Neonates 5 to 14 days old and sexually active
teens: C. trachomatis
Erythema, chemosis,
clear or
mucoid
exudate,
palpebral
follicles
Cultures (ELISA, PCR), R/O
sexual activity
Neonates: Erythromycin or
azithromycin PO
Adolescents: Doxycycline,
azithromycin, EES,
erythromycin base,
levofloxacin PO
Viral
conjunctivitis
Adenovirus 3, 4, 7; HSV, herpes zoster,
varicella
Erythema, chemosis,
tearing
(bilateral);
HSV and
herpes zoster:
unilateral with
photophobia,
fever; zoster:
nose lesion;
spring and fall
Cultures, R/O corneal
infiltration
Refer to ophthalmologist if
HSV or photophobia
present
Cool compresses three or
four times a day
Allergic and
vernal
conjunctivitis
Atopy sufferers, seasonal Stringy, mucoid
exudate,
swollen eyelids
and
conjunctivae,
itching (key
finding),
tearing,
palpebral
follicles,
headache,
rhinitis
Eosinophils in conjunctival
scrapings
Naphazoline/pheniramine,
naphazoline/antazoline
ophthalmic solution (see
text)
Mast cell stabilizer (see text)
Refer to allergist if needed
Otitis Externa
Otitis externa (OE), commonly called swimmer's ear, is a diffuse inflammation of the EAC and can involve the pinna or TM.
Inflammation is evidenced as (1) simple infection with edema, discharge, and erythema; (2) furuncles or small abscesses that
form in hair follicles; or (3) impetigo or infection of the superficial layers of the epidermis. OE can also be classified as mycotic
otitis externa, caused by fungus, or as chronic external otitis, a diffuse low-grade infection of the EAC. Severe infection or
systemic infection can be seen in children who have diabetes mellitus, are immunocompromised, or have received head and
neck irradiation.OE results when the protective barriers in the EAC are damaged by mechanical or chemical mechanisms. OE is most
frequently caused by retained moisture in the EAC, which changes the usually acidic environment to a neutral or basic
environment, thereby promoting bacterial or fungal growth. Chlorine in swimming pools adds to the 743problem because it kills
the normal ear flora, allowing the growth of pathogens. Regular cleaning of the EAC removes cerumen, which is an important
barrier to water and infection. Soapy deposits, alkaline drops, debris from skin conditions, local trauma, sweating, allergy,
stress, and hearing aids can also be responsible for causing OE (Rosenfeld et al, 2014).
OE is most often caused by Pseudomonas aeruginosa and Staphylococcus aureus, but it is not uncommon for the infection to be
polymicrobial. Furunculosis of the external canal is generally caused by S. aureus and Streptococcus pyogenes. Otomycosis is
caused by Aspergillus or Candida and can be the result of systemic or topical antibiotics or steroids. Otomycosis is also more
common in children with diabetes mellitus or immune dysfunction and in these cases is most commonly caused by Aspergillus
niger, Escherichia coli, or Klebsiella pneumonia. Group B streptococci are a more common cause in neonates.
Long-standing ear drainage may suggest a foreign body, chronic middle ear pathology (such as, a cholesteatoma), or
granulomatous tissue. Bloody drainage may indicate trauma, severe otitis media, or granulation tissue. Chronic or recurrent OE
may result from eczema, seborrhea, or psoriasis. Eczematous dermatitis, moist vesicles, and pustules are seen in acute infection,
whereas crusting is more consistent with chronic infection.
Clinical Findings
History
The following can be found:
• Itching and irritation
• Pain that seems disproportionate to what is seen on examination
• Pressure and fullness in ear and occasionally hearing loss that can be conductive or sensorineural
• Rare hearing loss and otorrhea or systemic complaints and symptoms
• Sagging of the superior canal, periauricular edema, and preauricular and postauricular lymphadenopathy with more severe disease
Extension to the surrounding soft tissue results in the obstruction of the canal with or without cellulitis.
Physical Examination
Findings on physical examination can include the following:
• Pain, often quite severe, with movement of the tragus (when pushed) or pinna (when pulled) or on attempts to examine the ear with an
otoscope
• Swollen EAC with debris, making visualization of the TM difficult or impossible
• Rare otorrhea
• Occasional regional lymphadenopathy
• Tragal tenderness with a red, raised area of induration that can be deep and diffuse or superficial and pointing, which is characteristic of
furunculosis
• Red, crusty, or pustular spreading lesions
• Pruritus associated with thick otorrhea that can be black, gray, blue-green, yellow, or white, and black spots over the TM are indicative of
mycotic infection
• Dry-appearing canal with some atrophy or thinning of the canal and virtually no cerumen visible with chronic OE
• Presence of pressure-equalizing tube or perforation of TM
Diagnostic Studies
Culturing the discharge from the ear is not customary but may be indicated if clinical improvement is not seen during or after treatment,
severe pain persists, the child is a neonate, the child is immunocompromised, or chronic or recurrent OE is suspected. Culturing requires a
swab premoistened with sterile nonbacteriostatic saline or water.
Differential Diagnosis
AOM with perforation, TTO, chronic suppurative otitis media (CSOM), necrotizing OE, cholesteatoma, mastoiditis, posterior auricular
lymphadenopathy, dental infection, and eczema are all possible differential diagnoses.
Management
The following steps outline the management of OE:
• Eardrops are the mainstay of therapy for OE (see Table 30-3). Eardrops containing acetic acid or antibiotic with and without corticosteroid
drops are the treatment of choice for OE. Symptoms should be markedly improved within 7 days, but resolution of the infection may takeup to 2 weeks. Drops should be used until all symptoms have resolved. Ototoxic drugs should not be used if there is a risk of TM
perforation.
• Antibiotic agents should be chosen based on efficacy, resistance patterns, low incidence of adverse effects, cost, and likelihood of
compliance. Neomycin, polymyxin, or hydrocortisone drops should not be used if the TM is not intact, because these drugs are
known to cause damage to the cochlea (Rosenfeld et al, 2014).
• The quinolone products are effective against Pseudomonas, S. aureus, and Streptococcus pneumoniae, which may be a factor if the OE
is a complication of AOM.
• Systemic antibiotics should not be used unless there is extension of infection beyond the ear or host factors that require more systemic
treatment (severe OE, systemic illness, fever, lymphadenitis, or failed topical treatment).
• Treatment for OE must include thorough parent education regarding the instillation of otic drops so that they are effective in eradicating
infection. The drops should be administered with the child lying down with the affected ear upward. Drops should run into the EAC until it
is filled. Move the pinna in a to-and-fro movement or pump the tragus to remove any trapped air and ensure filling (Rosenfeld et al, 2014).
The child should remain lying down for 3 to 5 minutes, leaving the ear open to the air.
744
• If the infection is severe and not improving in the first 5 to 7 days, aural irrigation with water, saline, or hydrogen peroxide may be tried, or
refer to the otolaryngologist for débridement and suction.
• If significant swelling is present, inserting a wick into the EAC is helpful. A wick made of compressed cellulose, hydrogel polymer
(Merocel XL), or gauze (0.25 inch) usually works well. The tip of the wick is lubricated with water or saline just before insertion into the
ear. Once in place, the wick should be impregnated with antibiotics for as long as it remains in the auditory canal. (This may require
reapplication of drops every 2 to 3 hours.) Wicks are usually removed after several days. The wick will fall out when the swelling has
subsided, and treatment with direct application of drops to the ear canal should continue for the entire course.
• Avoid cleaning, manipulating, and getting water into the ear. Swimming is prohibited during acute infection.
• Administer analgesics for pain. Narcotic analgesics may be necessary for severe pain but are only indicated for short-term use.
• Débridement with a cotton-tipped applicator, self-made cotton wick, or calcium alginate swabs is indicated once the inflammatory process
has subsided and can enhance the effectiveness of the ototopical antibiotic drops. Lance a furuncle that is superficial and pointed with a 14-
gauge needle. If it is deep and diffuse, a heating pad or warm oil-based drops can speed resolution.
• If impetigo is present, clear the canal by using water or an antiseptic solution followed by a warm-water rinse. Apply an antibiotic ointment
(mupirocin) twice a day for 5 to 7 days. There is increasing resistance to mupirocin, and retapamulin might be necessary in children over 9
months of age (Bangert et al, 2012; Drucker, 2012). The child should avoid touching the ear. Fingernails should be short, and hands should
be cleansed with soap and water. Systemic antibiotics are generally unnecessary.
• Fungal OE is uncommon in primary OE. Fungal OE is more likely related to chronic OE or following treatment with topical and/or systemic
antibiotics. Aspergillus and Candida species are most commonly seen in mycotic OE (Rosenfeld et al, 2014). Treatment consists of
antifungal solutions, such as clotrimazole-miconazole, nystatin, or other antifungal agents, including gentian violet and thimerosal 1 : 1000.
• The canal should be cleansed with a 5% boric acid in ethanol solution prior to antifungal solution.
If the child is not improved within 72 hours (relief of otalgia, itching, and fullness), recheck to confirm diagnosis. Lack of improvement
may be due to obstructed ear canal, foreign body, poor adherence, or contact sensitivity among other things. A follow-up visit may be
necessary after 1 to 2 weeks for reevaluation of the OE and removal of debris. If symptoms are worsening or there is no improvement in a
week, a referral to an otolaryngologist or dermatologist is indicated.
Complications
Infection of surrounding tissues with impetigo, irritated furunculosis, and malignant OE with progression and necrosis caused
by Pseudomonas are possible complications. Involvement of the parotid gland, mastoid bone, and infratemporal fossa is rare (Rosenfeld et al,
2014).
Prevention
The patient should be instructed to do the following:
• Avoid water in the ear canals.
• Use well-fitting earplugs for swimming especially in ―dirty water.‖
• Use alcohol vinegar otic mix (two parts rubbing alcohol, one part white vinegar, and one part distilled water) 3 to 5 drops daily, especially
after swimming or bathing, to prevent the recurrence of OE (Waitzman, 2015).
• Use a blow dryer on warm setting to dry the EAC.
• Avoid persistent scratching or cleaning of the external canal.
• Avoid prolonged use of ceruminolytic agents.
Hand-foot-mouth disease: This is a clinical entity evidenced by fever, vesicular eruptions in the oropharynx that may ulcerate,
and a maculopapular rash involving the hands and feet. The rash evolves to vesicles, especially on the dorsa of the hands and
the soles of the feet, and lasts 1 to 2 weeks (Fig. 24-1).Pharyngitis
Acetaminophen or ibuprofen
Antibiotics if GABHS
Saltwater gargles
Anesthetic lozenges for older child
Streptococcal Disease
Streptococci are gram-positive spherical cocci that are broadly classified based on their ability to hemolyze RBCs. Complete hemolysis is
known as beta-hemolytic. Partial hemolysis is alpha-hemolytic; non-hemolysis is gamma-hemolytic. Cell wall carbohydrate differences
further subdivide the streptococci. These differences are identified as Lancefield antigen subgroups A-H and K-V. Subgroups A-H and K-O
are associated with human disease. Group A beta-hemolytic streptococcus is the most virulent, although group B beta-hemolytic
streptococcus can cause bacteremia and meningitis in infants younger than 3 months old (rarely older). Group A streptococcus (GAS) are
also subdivided into more than 100 subtypes based upon their M protein antigen located on the cell surface and fimbriae on the cell's 535outer
edge. The virulence of GAS is greatly dependent upon their M protein. If the M protein is present, GAS strains are able to resist
phagocytosis; if the M protein is weak or absent, the strains are basically avirulent (e.g., chronic GAS pharyngeal carriers). GAS also
produces many varieties of enzymes and toxins that may stimulate specific antitoxin antibodies for immunity or serve as evidence of past
infection but not confer immunity. There may also not be cross-immunity between antibodies for different GAS strains (e.g., scarlet fever is
caused by three different pyrogenic exotoxins, so the illness can recur). Some general remarks about specific illnesses due to GAS and nongroup A and B streptococcus infection are discussed in this chapter; cross-references to specific chapters are noted for other GAS caused
infections.
Group A Streptococcus
Streptococcus microbes most commonly invade the respiratory tract, skin, soft tissues, and blood. Transmission is primarily through infected
upper respiratory tract secretions or, secondarily, through skin invasion. Fomites and household pets are not vectors. Food-borne outbreaks
from contamination by food handlers have been reported. Both streptococcus pharyngitis and impetigo are associated with crowding, whether
at home, school, or other institution. Streptococcal pharyngitis is rare in infants and children younger than 3 years old, but the incidence rises
with age and is most common in the winter and early spring in temperate climates when respiratory viruses circulate. Carrier rates in
asymptomatic children are up to 20% (Arnold and Nizet, 2012). By contrast, streptococcus skin infection (impetigo, pyoderma) is more
common in toddlers and preschool-age children. Those at increased risk for invasive GAS are individuals with varicella infection, IV drug
use, HIV, diabetes, chronic heart or lung disease, infants, and older adults.
The incubation period is 2 to 5 days for pharyngitis and 7 to 10 days from skin acquisition to development of impetiginous lesions. In
untreated individuals, the period of communicability is from the onset of symptoms up to a few months. Children are generally considered
non-infectious 24 hours after the start of appropriate antibiotic therapy.
Clinical Findings and Diagnostic Studies
The following may be seen in GAS:
• Respiratory tract infection: Streptococcal tonsillopharyngitis (GABHS) and pneumonia are described in Chapter 32. Peritonsillar abscess,
cervical lymphadenitis, retropharyngeal abscess, otitis media, mastoiditis, and sinusitis symptoms may be clinical features.
• Scarlet fever: This is caused by erythrogenic toxin. It is uncommon in children younger than 3 years old. The incubation period is
approximately 3 days (the range is 1 to 7 days). There is abrupt illness with sore throat, vomiting, headache, chills, and malaise. Fever can
reach 104° F (40° C). Tonsils are erythematous, swollen, and usually covered in exudate. The pharynx also is inflamed and can be covered
with a gray-white exudate. The palate and uvula are erythematous and reddened, and petechiae are present. The tongue is usually coated and
red. Desquamation of the coating leaves prominent papillae (strawberry tongue). The typical scarlatina rash appears 1 to 5 days following
onset of symptoms but may be the presenting symptom. The exanthema is red, blanches to pressure, and is finely papular, making the skin
feel coarse, with a sandpaper feel. The rash generally begins on the neck and spreads to the trunk and extremities becoming generalized
within 24 hours. The face may be spared (cheeks may be reddened with circumoral pallor), but the rash becomes denser on the neck, axilla,
and groin. Pastia lines, transverse linear hyperpigmented areas with tiny petechiae, are seen in the folds of the joints (see Fig. 24-3). In
severe disease, small vesicles (miliary sudamina) can be found on the hands, feet, and abdomen. There is circumoral pallor and the cheeks
are erythematous. The rash begins to fade and desquamate after 3 to 4 days starting on the face and slowly moving to the trunk and
extremities and may include fingernail margins, palms, and soles; this process can take up to 6 weeks. Sore throat and constitutional
symptoms resolve in approximately 5 to 7 days (average 3 to 4 days).
• Bacteremia: This can occur after respiratory (pharyngitis, tonsillitis, AOM) and localized skin infections. Some children have no obvious
source of infection. Meningitis, osteomyelitis, septic arthritis, pyelonephritis, pneumonia, peritonitis, and bacterial endocarditis are rare but
are associated with GAS bacteremia. (Neonatal sepsis due to group B streptococcus is discussed in Chapter 39.)
• Vaginitis and streptococcal toxic shock syndrome (see discussions in Chapter 36).
• Perianal streptococcal cellulitis: Symptoms include local itching, pain, blood-streaked stools, erythema, and proctitis. Fever and systemic
infections are uncommon. Although infection is usually the result of autoinoculation, sexual molestation is in the differential.
• Skin infections (see Chapter 37); rheumatic heart disease (see Chapter 25); and necrotizing fasciitis (see Chapter 37).
Refer to disease-specific chapters for diagnostic studies of disease-specific conditions.Differential Diagnosis, Management, and Complications
Many viral pathogens are on the differential for acute pharyngitis, including influenza, parainfluenza, rhinovirus, coronavirus, adenovirus,
and respiratory syncytial virus. EBV is common and is usually accompanied by other clinical findings (e.g., splenomegaly, generalized
lymphadenopathy). Other causes of bacterial upper respiratory disease include (though rare) diphtheria, tularemia, toxoplasmosis,
mycoplasma, tonsillar TB, salmonellosis, and brucellosis (Gerber, 2011). Staphylococcal impetigo must be differentiated from GABHS
pyoderma. Septicemia, meningitis, osteomyelitis, septic arthritis, pyelonephritis, and bacterial endocarditis can result from other bacteria
causing similar infections.
536
Antimicrobial therapy is recommended for GABHS-caused pharyngitis to decrease the risk of acute rheumatic fever, decrease the length
of the illness, prevent complications, and reduce transmission to others. See appropriate aforementioned site-specific chapters for
recommendations for managing specific infections.
Complications are usually caused by the spread of the disease from the localized infection. Upper respiratory complications include
cervical lymphadenitis, retropharyngeal abscess, otitis media, mastoiditis, and sinusitis if the primary infection is unrecognized or treatment
is inadequate. Acute poststreptococcal glomerulonephritis can occur following skin or upper respiratory GAS infection, whereas acute
rheumatic fever only occurs following GAS URIs. Poststreptococcal reactive arthritis can occur following GAS pharyngitis. Skin infection
with GAS may progress to cellulitis, myositis, or necrotizing fasciitis. Other complications may be associated with invasive infections
including pneumonia, pleural empyema, meningitis, osteomyelitis, and bacterial endocarditis.
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) is a group of neuropsychiatric
disorders thought to result from the production of autoimmune antibodies; these include obsessive-compulsive disorders, tic disorders, and
Tourette syndrome. See Chapter 19 for further discussion.
Non–Group A or B Streptococci
These streptococci or Lancefield groups (principally groups C and G) are associated with invasive disease in all age groups. They may cause
septicemia, UTIs, endocarditis, respiratory disease (upper and lower), skin soft tissue infection, pharyngitis, brain abscesses, and meningitis
in newborns, children, adolescents, and adults. The incubation period and communicability times are unknown. Positive culture from
normally sterile body fluids is adequate for diagnosis. Penicillin G is the drug of choice with modification based on culture sensitivities.
Pneumonia with empyema or abscess may respond slowly despite effective antimicrobial therapy with fevers lasting more than 7 days
(Haslam and St. Geme, 2012).
Kawasaki Disease
KD (also known as mucocutaneous lymph node syndrome or infantile polyarteritis) is the second most common 563childhood
vasculitis with a varying incidence from country to country, with Japan having the highest incidence of 239.6 per 100,000. The
incidence is increasing in Japan, the United Kingdom, and India (Saundankar et al, 2014). The disease is characterized by an
acute generalized systemic medium vessel vasculitis occurring throughout the body. Although its cause is unknown, it is
believed that an infectious agent activates the immune system in a genetically susceptible host. Genetics may explain the higher
incidence in Asia as well as a higher incidence in children of parents or siblings with a history of the disease. Recent data
suggest T-cell activation plays a role in disease severity and susceptibility (Scuccimarri, 2012).
KD exhibits geographic and seasonal outbreaks, in the late winter and early spring. Person-to-person spread is low. Referral
of these children to a pediatrician is necessary. It is self-limited and the most common cause of acquired heart disease in
children in Japan and the United States (Saundankar et al, 2014). The EULAR/PReS classification for KD includes a persistent
fever for at least 5 days plus four of the following (Ozen et al, 2010):
• Bilateral conjunctival injection
• Changes of the lips and oral cavity
• Cervical lymphadenopathy
• Polymorphous exanthema
• Changes in the peripheral extremities (swelling of the hands or feet) or perineal area
Clinical Findings
Despite accepted KD guidelines, children can have atypical or incomplete KD with coronary anomalies shown by echocardiogram. Children
younger than 6 to 12 months old may have more atypical findings. In atypical KD, the child may fulfill the criteria but has an additional
feature that is not usually seen in KD. In incomplete KD, the fever may last for 5 days or more, but the child will only meet two or three of
the other criteria. Incomplete KD is more common in children younger than 1 year old and older than 9 years old. Thus, incomplete KD
without nodal involvement is possible. Coronary artery involvement is found more frequently in children with incomplete KD, so based on
the frequency of the disease, an index of suspicion should be maintained in infancy and older school-age children (Scuccimarri, 2012). If KD
is untreated, the normal course of fever is 10 to 14 days.
Other clinical features associated with KD include irritability, aseptic meningitis, mild acute iridocyclitis or anterior uveitis, otitis media
due to inflammation rather than infection of the drum, urethritis, hydrops of the gallbladder, and facial nerve palsy. In children who havereceived BCG, there may be erythema and induration at the site of injection. Two rare complications are MAS and peripheral gangrene
(Scuccimarri, 2012).
Stage 1: Acute Phase
The acute phase (days 0 to 14) begins with an abrupt onset of high fever (greater than 102.2° F [39° C]) that is unresponsive to antipyretics or
antibiotics. Significant irritability, bilateral nonpurulent conjunctival injection, erythema of the oropharynx, dryness and fissuring of the lips,
―strawberry tongue,‖ cervical lymphadenopathy, a polymorphous rash, erythema of the urethral meatus, tachycardia, and edema of the
extremities are typically noted. During the acute phase, there may be pericardial, myocardial, endocardial, and coronary artery inflammation.
The child typically is tachycardic and has a hyperdynamic precordium with a gallop rhythm and a flow murmur. Rarely, children have low
cardiac output syndrome from poor myocardial function.
Stage 2: Subacute Phase
The subacute phase (2 to 4 weeks after illness onset) begins with resolution of the fever and lasts until all other clinical signs have
disappeared. Irritability may be prolonged throughout this phase. Desquamation of the fingers (at the junction of nail tip and digit) occurs
first, followed by desquamation of the toes. Transient jaundice, abnormal liver function tests, arthralgia or arthritis, transient diarrhea,
orchitis, facial palsy, and sensorineural hearing loss may occur. Coronary artery aneurysms appear during this period—more so in untreated
children. Common sites for aneurysms, in order of frequency, are the proximal left anterior descending coronary, proximal right coronary,
left main coronary, left circumflex, and distal right coronary artery.
Stage 3: Convalescent Phase
During the convalescent phase, all clinical signs of KD have resolved, but laboratory values may not have returned to normal. This phase is
complete when all blood values are normal (6 to 8 weeks from onset). However, nail changes including Beau lines (deep transverse grooves
across the nails) may be seen (Scuccimari, 2012).
Although some researchers note a chronic phase lasting from 40 days to years after illness onset, this phase is not present in all patients.
Although coronary complications, if present, can persist into adulthood, a recent study of 564 patients with KD revealed a low incidence of
side effects in children who were followed to 21 years of age (Holve et al, 2014).
Diagnostic Studies
KD is a diagnosis of exclusion. Results of lab investigations are not diagnostic but rather help rule in other diagnoses. Although the acute
phase reactants (ESR and CRP) are usually increased, they may be normal early in the course of the illness. A CBC may show an increased
WBC with a predominance of neutrophils with toxic granulation. Anemia may follow with prolonged inflammation. A marked
thrombocytosis with values greater than 1 million follow in the second week of illness in the subacute phase. The comprehensive metabolic
profile may show an increase in serum transaminases and hypoalbuminemia. Sterile pyuria may occur. Leukopenia and thrombocytopenia
in 564KD may occur in association with the life-threatening MAS.
• Stage 1 is typified by an elevated ESR and platelet count (as high as 700,000/mm3), elevated CRP, leukocytosis with left shift, slight
decreases in red blood cells and hemoglobin, hypoalbuminemia, increased α2-globulin, and sterile pyuria. The platelet count may be initially
normal with gradual increase after the seventh day of fever.
• Blood, urine, cerebrospinal fluid, and group A beta-hemolytic streptococci (GABHS) pharyngeal cultures may be indicated given the
symptomatology (to rule out other sources of fever).
• Echocardiograms at acute illness, 2 weeks and 6 to 8 weeks after onset of fever, are performed to evaluate for coronary, myocardial, and
pericardial inflammation. Angiography, MRI, and cardiac stress testing may be considered.
Differential Diagnosis
The differential diagnosis includes viral infections (e.g., measles, adenovirus, EBV, enterovirus, influenza, or roseola) and bacterial infections
(e.g., cervical adenitis, scarlet fever, staphylococcal scalded skin syndrome, toxic shock syndrome, leptospirosis, or Rickettsia illness, such as
Rocky Mountain spotted fever). Immune-mediated diseases may need to be considered and include Steven-Johnson syndrome, serum
sickness, RF, SJIA or other JIA, or connective tissue diseases, such as SLE. Other differential diagnoses include mercury poisoning, or tumor
necrosis factor receptor–associated periodic syndromes, such as hyper IgM syndrome (Scuccimarri, 2012).
Management
• Early diagnosis is essential to prevent aneurysms in the coronary and extraparenchymal muscular arteries. Treatment goals include: (1)
evoking a rapid anti-inflammatory response, (2) preventing coronary thrombosis by inhibiting platelet aggregation, and (3) minimizing
long-term coronary risk factors by exercise, a heart healthy diet, and smoking prevention. The child should be referred for initial treatment
that includes the following medications and agents (Scuccimarri, 2012):
• Intravenous immunoglobulin (IVIG) therapy (a single dose of 2 g/kg over 12 hours, ideally in the first 10 days of the illness) to reduce
the incidence of coronary artery abnormalities. The use of immunoglobulin after the tenth day must be individualized. If a child is
found to have an abnormal echocardiogram, fever, tachycardia, or other signs of inflammation beyond the tenth day, then
immunoglobulin is still indicated. Retreatment with immunoglobulin may be useful for persistent or recurrent fevers.
• High-dose aspirin is given for its anti-inflammatory properties (80 to 100 mg/kg/day in four divided doses—every 6 hours initially)
until afebrile for at least 48 to 72 hours, then lowering the aspirin dose to 3 to 5 mg/kg/day until 6 to 8 weeks and then candiscontinue if the echocardiogram is normal. If significant coronary artery abnormalities develop and do not resolve, aspirin or other
antiplatelet therapy is used indefinitely.
• For patients with IVIG-resistant disease as indicated by a persistent fever 48 hours after treatment with IVIG and aspirin, a second
treatment of IVIG at 2 mg/kg over 12 hours is initiated. If this is not successful, then methylprednisone IV at 30 mg/kg over 3 hours
once a day for 1 to 3 days may be initiated. Infliximab 5 mg/kg may also be used. If the patient is still febrile, then the opposite antiinflammatory can be used. (Methylprednisone in the infliximab groups, or infliximab in the methylprednisone group.) Other options
include cyclosporine A, methotrexate or cyclophosphamide (Saneeymehri et al, 2015).
• An echocardiogram should be obtained as soon as the diagnosis is established as a baseline study, with subsequent studies at 2 weeks and 6
to 8 weeks after onset of illness. If a child is found to have abnormalities, more frequent evaluations may be indicated.
• All children on chronic aspirin therapy should receive inactivated influenza vaccination. If varicella or influenza develops, aspirin treatment
should be stopped for 6 weeks and another antiplatelet drug substituted to minimize the risk of Reye syndrome.
• Live virus vaccines should be delayed until 11 months after administration of IVIG (AAP Red Book, 2015).
• Children without coronary or cardiac changes should be followed by a cardiologist during the first year after the onset of KD. If there are
no cardiac changes during that first year, then the PCP may follow the patient with no activity restrictions imposed at that point.
• Patients with any range of transient coronary artery dilation (including giant aneurysms) should be followed by a cardiologist for years;
physical activity limitations may be imposed.
• Follow and counsel all KD patients about a heart-healthy diet.
Complications and Prognosis
The acute disease is self-limited; however, during the initial stage (acute phase), inflammation of the arterioles, venules, and capillaries of the
heart occurs and can later progress to coronary artery aneurysm in 15% to 25% of untreated children (less than 5% when treated
appropriately). The process of aneurysm formation and subsequent thrombosis or scarring of the coronary artery may occur as late as 6
months after the initial illness. Other possible complications include recurrence of KD (less than 2%); CHF or massive myocardial infarction;
myocarditis or pericarditis, or both (30%); pericardial effusion; and mitral valve insufficiency. Mortality (1.25%) from KD occurs from
cardiac sequelae 15 to 45 days after onset of fever. Children with coronary dilation or aneurysms (especially those greater than 4 mm) may
have long-term coronary endothelial changes that place the child at risk for early ischemic disease; 565they may also develop dyslipidemias
(Wood and Tulloh, 2009). Studies from Japan raise concern about risk of early atherosclerosis (due to arterial damage, ongoing inflammatory
process, and alteration in lipid profile and other atherosclerosis risk factors) even in children without coronary changes during acute febrile
illness (Fukazawa and Ogawa, 2009).
The risk of coronary aneurysm is reduced in patients older than 1 year old if IVIG is given within 10 days of the illness. Aneurysm
regression occurs in half of all patients who develop them, commonly by 1 year after the illness (80% resolve within 5 years), but vessels do
not dilate normally in response to increased oxygen demand by the myocardium. Prompt treatment of chest pain, dyspnea, extreme lethargy,
or syncope is always warranted. Surgical revascularization and transcatheter revascularization are used for some coronary sequelae of KD
(Wood and Tulloh, 2009).
Acute Rheumatic Fever
ARF is a nonsuppurative complication following a Lancefield GAS pharyngeal infection that results in an autoimmune inflammatory process
involving the joints (polyarthritis), heart (rheumatic heart disease), CNS (Sydenham chorea), and subcutaneous tissue (subcutaneous nodules
and erythema marginatum). Recurrent ARF with its multisystem responses can follow with subsequent GAS pharyngeal infections. Longterm effects on tissues are generally minimal except for the damage done to cardiac valves that leaves fibrosis and scarring and results in
rheumatic heart disease. ARF is diagnosed based on a set of criteria called the revised Jones criteria (1992). These criteria are used for the
initial attack of ARF. Further modifications of the Jones criteria are used for recurrent ARF.
Clinical Findings and History
The diagnosis of an initial attack of ARF is based on the following revised Jones criteria:
• Evidence of documented (culture, rapid streptococcal antigen test, or ASO titer) GAS pharyngeal infection
• Findings of two major manifestations or one major and two minor manifestations of ARF (Berard, 2012; Burke and Chang, 2014)
Major Manifestations
Children with fewer manifestations can also have ARF. Arthritis of large joints occurs in 65% of cases, carditis in 50%, chorea in
15% to 30%, cutaneous nodules in 5%, and subcutaneous nodules in less than 7%. There is some controversy regarding the use
of the Jones criteria in developing countries where the ability for diagnostic testing may be limited; therefore, the World Health
Organization (WHO) criteria (Box 25-2) may be used (Ferrieri, 2002; Seckel and Hoke, 2011).
• Carditis is common (pancarditis, valves, pericardium, myocardium) and can cause chronic, life-threatening disease (i.e.,
congestive heart failure [CHF]) with estimates of 30% to 80% of patients with ARF experiencing carditis; it is more common in
younger children than adolescents. The symptoms of carditis may be vague and insidious with decreased appetite, fatigue,
and pains. A high-pitched holosystolic murmur is heard at the apex with radiation to the infrascapular area, as well as
tachycardia and often a gallop rhythm. Mitral and possibly aortic regurgitation occur in 95% of cases, usually within 2 weeksof RF illness. The mitral valve becomes leaky due to annular dilation and elongation of the chordate that attach leaflets to the
left ventricle. With moderate to severe mitral regurgitation CHF develops; recurrent episodes of RF lead to worsening valve
disease.
• Polyarthritis (migratory and painful) involving large joints and rarely small or unusual joints (e.g., vertebrae); it is the most
common manifestation of ARF.
• Sydenham chorea is uncommon.
560
• Erythema marginatum manifested as pink macules on the trunk and extremities; nonpruritic; this sign is uncommon.
• Subcutaneous nodules associated with repeated episodes and severe carditis; this sign is uncommon.
Minor Manifestations
• Fever (101° F to 102° F [38.2° C to 38.9° C]), arthralgia, history of ARF
Diagnostic Studies
• Elevated acute-phase reactants (ESR, white blood cells [WBCs], CRP)
• Leukocytosis
• Prolonged PR interval on ECG
Children may be diagnosed with ARF without evidence of a preceding streptococcal infection in the following two situations: (1) a child
with Sydenham chorea or (2) with acquired heart disease (commonly mitral valve regurgitation without a congenitally abnormal or prolapsed
valve) that can only be linked to ARF. Approximately 80% of children with ARF have an elevated ASO titer. A combination of both DNaseB testing and ASO rising may confirm the recent infection.
Differential Diagnosis
ARF is a clinical diagnosis associated with rising antibody titers. Arthritis and arthralgia can accompany a variety of diseases including JIA;
connective tissue diseases; viral infections, such as parvovirus; inflammatory bowel disease; bacterial infections, such as gonorrhea;
hemophilia; infective endocarditis; and Lyme disease (Berard, 2012). A complete history and physical examination with appropriate
diagnostic testing are critical to establish the diagnosis.
Management
The treatment of ARF includes the following:
• Antibiotic therapy to eradicate GAS infection: Primary prevention requires that a GAS infection be treated within 10 days of onset.
Benzathine penicillin G is the drug of choice unless there is an allergic history; erythromycin is then the drug of choice. Azithromycin and
cephalosporins are also sometimes used (Gerber, 2011). A patient with a history of ARF who has an upper respiratory infection should be
treated for GAS whether or not GAS is recovered as asymptomatic infection can trigger a recurrence.
• Anti-inflammatory therapy: Aspirin can be used for arthritis after the diagnosis is established; it is usually 561given only for 2 weeks and
then tapered. It is also used to treat mild to moderate carditis. Aspirin and steroids provide symptomatic relief but do not prevent the
incidence of chronic heart disease. Steroids have been beneficial in the management of severe carditis, reducing its morbidity and mortality.
The association of Reye syndrome with aspirin use is always a concern and must be addressed with parents. Yearly influenza immunization
is critical for children on aspirin therapy.
• Chest radiographs, ECG, and echocardiography are indicated; carditis usually develops within the first 3 weeks of symptoms.
• Referral for CHF treatment if needed: medical management and or valve replacement.
• Bed rest is generally indicated only for children with CHF. Children with Sydenham chorea may need to be protected from injury until their
choreiform movements are controlled. Steroids in the absence of other symptoms are not useful in the treatment of chorea.
• Children with severe chorea may benefit from the use of antiepileptic agents, such as sodium valproate or carbamazepine.
Prevention of Acute Rheumatic Fever
• Treat GAS pharyngeal infections with appropriate antibiotics. Antibacterial prophylaxis for those with a prior history of ARF is required
because of the greatly increased risk of recurrent ARF with subsequent inadequately treated GAS infections. Intramuscular penicillin G (1.2
million units) is more effective than daily penicillin V (Gerber, 2011) and must be given every 4 weeks (every 28 days) not monthly. It can
be given every 3 weeks in high-risk children.
• Antibacterial secondary prophylaxis with penicillin is given every 4 weeks for 5 years after the last ARF episode in children without carditis
or until 21 years old (whichever is longer). For those with carditis and persistent myocardial or valvular disease, treatment is 10 or more
years and may be lifelong (Gerber, 2011). In the majority of patients, valvular disease will resolve if they are compliant in taking antibiotic
prophylaxis after the first episode of rheumatic heart disease.
ComplicationsChronic CHF can occur after an initial episode of ARF or follow recurrent episodes of ARF. Residual valvular damage is responsible for
CHF. The risk of significant cardiac disease increases dramatically with each subsequent episode of ARF; thus prevention of subsequent
GAS infections is critical. Engagement in the follow-up is essential to prevent the need for cardiac valvular repair.
Bronchiolitis
Bronchiolitis is also called infectious asthma, asthmatic bronchitis, wheezy bronchitis, or virus-induced asthma. Bronchiolitis is a
disease that causes inflammation, necrosis, and edema of the respiratory epithelial cells in the lining of small airways, as well as
copious mucus production (Ralston et al, 2014). Bronchiolitis is characterized by the insidious onset of URI symptoms over 2 to 3
days that progresses to lower respiratory symptoms that last as long as 10 days (Da Dalt et al, 2013). It is a communicable
disease found primarily in infancy to 2 years old (Teshome et al, 2013) that accounts for 10% of visits to a primary provider the
first 2 years of life (Schroeder and Mansbach, 2014). Bronchiolitis is a common diagnosis used for an infant seen with wheezing
for the very first time and is the leading cause of hospitalizations for infants. The most common age for severe disease occurs in
infants between 2 to 3 months due to the natural postnatal nadir in maternal immunoglobulins received via the placenta during
the last trimester (Da Dalt et al, 2013). More than 80% of the cases of bronchiolitis occur in infants younger than 1 year of age
with a male-to-female ratio of 1.5 : 1 (Welliver, 2009). In mild cases, symptoms can last for 1 to 3 days. In severe cases, cyanosis,
air hunger, retractions, and nasal flaring with symptoms of severe respiratory distress within a few hours may be seen. Apnea
can occur with a wide range of prevalence reported (Ralston et al, 2014) and may require mechanical ventilation.
Newer understanding of the pathophysiology in bronchiolitis points to airway obstruction as a result of epithelial and
inflammatory cellular debris due to infiltration of the virus into the small bronchiole epithelium and alveolar epithelial cells
(AEC), types I and II. Membranous pneumatoceles, or AEC type I, are dominant and cover 96% of the respiratory tree. Their role
is in gas exchange, whereas AEC type II are important to surfactant production (Chuquimia et al, 2013). It is a disease of the
small bronchioles that are 2 mm in size. There is a sparing of basal cells in the bronchiole. The main lesion is epithelial necrosis,
which leads to a dense plugging of the bronchial lining. This results in increased airway resistance, atelectasis, hyperinflation,
and increased mucus production (Teshome et al, 2013).
Bronchiolitis is a viral illness predominantly caused by RSV, especially in outbreaks (Da Dalt et al, 2013; Welliver, 2009).
Recent data suggest that up to 30% of infants with severe bronchiolitis are co-infected with two or more viruses (Mansbach et al,
2012). In descending order after RSV, rhinovirus, parainfluenza, adenovirus, and mycoplasma are causes (Teshome et al, 2013).
Metapneumovirus was discovered in 2001 and is a cause of bronchiolitis 7% of the time. Human bocavirus is a common coinfecting virus with RSV and is found up to 80% of the time (Teshome et al, 2013). RSV-specific immunoglobulin E (IgE),
eosinophils, and chemokines may play a role in the pathogenesis of bronchiolitis (Welliver, 2009). Adenovirus and RSV can
cause long-term complications. The incubation period for RSV is 2 to 8 days and typically occurs from November through
March with virtually no outbreaks in the summer (Teshome et al, 2013; Welliver, 2009). Fever tends to be higher with
adenovirus versus RSV (Teshome et al, 2013).
Respiratory viruses are spread by close contact with infected respiratory secretions or fomites and can live on 818surfaces for
up to 30 minutes (Teshome et al, 2013). The most frequent mode of transmission is hand carriage of contaminated secretion. The
source of infection is an older child or adult family member with a “mild” URI. Older children and adults have larger airways
and tolerate the swelling associated with this infection better than infants do. Most cases of bronchiolitis resolve completely, but
recurrence of infection is common, and symptoms tend to be mild.
Infants who are at higher risk of severe RSV include children with major chronic pulmonary disease, such as CF,
neuromuscular disorders, or bronchopulmonary dysplasia; premature birth before 35 weeks of gestational age; and infants with
significant hemodynamically difficulties due to congenital heart disease (Teshome et al, 2013). Other risk factors for severe RSV
disease are male gender, crowded household, lack of breastfeeding, smoke exposure, day care attendance, having siblings, birth
during the winter months, and immunodeficiency (Da Dalt et al, 2013).
Clinical Findings
History
The following are reported:
• Initial presentation: Typically the illness begins with URI symptoms of cough, coryza, and rhinorrhea and progresses over 3 to 7 days
(Smith, 2011).
• Gradual development of respiratory distress marked by noisy, raspy breathing with audible expiratory wheezing.
• Low-grade to moderate fever up to 102° F (38.9° C).
• Decrease in appetite.
• No prodrome in some infants; rather they have apnea as the initial symptom.• Usually the patient's course is the worst by 48 to 72 hours after the wheezing starts and then the patient starts to improve. If the child has a
bacterial illness, the child will continue to worsen with a high fever.
Physical Examination
Findings include the following:
• Upper respiratory findings
• Coryza
• Mild conjunctivitis in 33% (Welliver, 2009)
• Pharyngitis
• Otitis media in up to 15% (Welliver, 2009)
• Lower respiratory findings (Teshome et al, 2013)
• Tachypnea (approximately 40 to 80 breaths per minute)
• Substernal and/or intercostal retractions
• Heterophonous expiratory wheezing
• Fine or coarse crackles may be heard throughout the breathing cycle
• Varying signs of respiratory distress and pulmonary involvement (e.g., nasal flaring, grunting, retractions, cyanosis, prolonged
expiration)
• Abdominal distention
• Palpable liver and spleen, pushed down by hyperinflated lungs and a flattened diaphragm
Diagnostic Studies
A diagnosis of bronchiolitis should be based on the history and physical examination (Ralston et al, 2014). Overuse of diagnostic testing
persists in clinical practice despite available guidelines on the diagnosis and management of bronchiolitis (Librizzi et al, 2014; Ralston et al,
2014; Turner et al, 2014). The routine use of chest radiographs in previously healthy infants with mild RSV bronchiolitis is not indicated.
Evidence-based guidelines from the AAP and the Scottish Intercollegiate Guidelines Network (SIGN) are strongly against routine chest
radiography, including those in previously healthy infants with mild RSV bronchiolitis (Ralston et al, 2014; SIGN, 2006). In severe illness, a
chest x-ray may be ordered to rule out pneumonia or pneumothorax, but its use must be weighed against the dangers of radiation exposure.
The findings of chest radiography can vary, and even with severe illness the x-ray can be clear with a flattened diaphragm and an increase in
anteroposterior diameter. Areas of atelectasis can appear like a pneumonitis, but true pneumonia is uncommon (early bacterial pneumonia can
be difficult to detect and cannot be ruled out by radiographs).
Routine virologic testing is not recommended. In selected situations (hospitalization or if an infant has received monthly palivizumab
[Synagis]), enzyme-linked immunosorbent assays or fluorescent antibody techniques to look for RSV are the diagnostic procedures of choice
in most laboratories. Viral culture of nasal washings can be done in severe cases to confirm RSV, parainfluenza viruses, influenza viruses,
and adenoviruses. PCR is helpful in deciding about isolation of cohorts with the same infection in the hospital setting. The cost of the
diagnostic viral testing may outweigh the clinical usefulness of knowing which virus is infecting the patient.
Hematologic testing is not recommended in the latest guidelines. If a CBC is done for another reason, a mild leukocytosis may be seen
with 12,000 to 16,000/mm3. Routine laboratory tests are usually not required to confirm the diagnosis, because they lack specificity.
However, young infants pose a diagnostic dilemma, because they are at greater risk of a serious bacterial infection (SBI) and, therefore, blood
cultures and CBC with differential are done with a higher rate of antibiotic use in infants who had these blood tests (Librizzi et al, 2014).
Urine cultures actually have a higher rate of positive results in the young febrile infant (up to 2.3% in a bronchiolitis study conducted by
Librizzi and colleagues).
Differential Diagnosis
The diagnosis of bronchiolitis can be confused with asthma, but there are some differences that may be helpful. Asthma is an acute process
due to airway hyperreactivity and inflammation, whereas the onset of bronchiolitis is insidious. The response to the usual asthma therapies of
beta agonist and 819steroids is poor in infants with bronchiolitis. In contrast, certain viral illnesses in young children can induce wheezing
that will respond to a β-agonist with good results.
FB aspiration is discussed in greater detail later in this chapter, but this is usually a toddler with a history of choking who then develops
focal areas of wheezing. Although children with congestive heart failure can wheeze, they also show symptoms of sweating and the signs of
failure to thrive with a murmur and an S4 gallop rhythm. Other differentials include airway irritants, gastroesophageal reflux, pneumonia,
allergic pneumonitis, vascular rings, lung cysts, and lobar emphysema (Teshome et al, 2013; Welliver, 2009).
Management
Evidence-based guidelines published by the AAP no longer support a trial of bronchodilators as an option for infants and children with
bronchiolitis because of the risk associated with its use and the lack of evidence of an effect (Ralston et al, 2014; Schroeder and Mansbach,
2014). The use of epinephrine is also not recommended for infants and children. Administration of nebulized hypertonic saline to infants in
the emergency department is not recommended; however, nebulized hypertonic saline can be administered to infants and children diagnosed
with bronchiolitis and hospitalized. Systemic corticosteroids should not be administered in the treatment of bronchiolitis in infants; chest
physiotherapy is contraindicated in infants and children. Antibiotics have no place in the treatment of a viral disease (such as, bronchiolitis),
unless there is a concomitant bacterial infection or strong suspicion. Most infants with mild signs of respiratory distress can be treated as
outpatients if their oxygen level is within a normal range (Ralston et al, 2014; Schroeder and Mansbach, 2014):
• Supportive care consists of adequate hydration and use of antipyretics.• The need for supplemental oxygen administration is based on oxyhemoglobin saturation levels. If an infant's or child's oxyhemoglobin level
is greater than 90%, the decision to administer oxygen is left up to the provider (Ralston et al, 2014).
• Transcutaneous oxygen saturation monitoring (continuous pulse oximetry) is also an individual provider's choice (Ralston et al, 2014).
• Fluid intake is strongly recommended to prevent dehydration.
• Nasal suctioning to clear the upper nasal passages is recommended.
The inpatient management of bronchiolitis may include using heated, humidified, high-flow oxygen via nasal canula. The mechanism of
action is to improve mucous ciliary clearance and avoid nasal dryness. The high flow delivers positive airway pressure to keep the alveoli
open and reduce ventilation perfusion mismatch and small airway microatelectasis (Da Dalt et al, 2013). This method needs more research
but is being regularly used in the inpatient basis (Schroeder and Mansbach, 2014; Teshome et al, 2013).
Hypertonic saline (3%) is being used to treat bronchiolitis in hospitalized infants and children. The mechanism of action is due to
decreasing mucus viscosity, thus improving airway clearance. It is not recommended for outpatient use and does not reduce hospital
admission in patients being treated in the emergency department. However, its use does reduce length of hospital stay (Da Dalt et al,
2013; Zhang et al, 2008). Research on this method is ongoing.
The use of deep airway suctioning is avoided, though continuing to keep the nasal airway clear on a regular basis may improve airflow.
This intervention is intuitive and does not need a randomized trial to show its benefit (Schroeder and Mansbach, 2014).
As stated previously, there is no evidence for the routine use of antibiotics, β-agonist, or corticosteroids. Ribavirin is no longer
recommended routinely and is presently only used in infants with severe illness due to underlying immunodeficiency, chronic lung disease,
or hemodynamically unstable cardiac conditions (Da Dalt et al, 2013). Although leukotriene levels are high in bronchiolitis, the use of
antileukotriene inhibitors has not been adequately studied and, thus, is not recommended. A recent review showed an increased risk of
bronchiolitis with low cord blood vitamin D level (Belderbos et al, 2011). At present, there is no evidence to show any pharmacologic
therapy is clearly superior.
Parents caring for infants and children at home need to understand:
• The management of rhinitis (use of saline drops and suctioning of nares)
• Indications for the use of antipyretics
• The use of home oxygen
• Signs of increasing respiratory distress or dehydration that call for hospitalization
• Guidelines for feeding an infant with signs of mild respiratory distress (amount of fluid needed per 24 hours; smaller, more frequent
feedings; monitoring of the respiratory rate; and guarding against vomiting)
• Education that infants and children with bronchiolitis typically have symptoms for 2 to 3 weeks
Infants younger than 2 months old and older infants with signs of severe respiratory distress should be hospitalized. Signs that suggest
increasing respiratory distress include the following (Smith, 2011):
• Progressive stridor or stridor at rest
• Apnea
• Increasing respiratory rate (sleeping rate of greater than 50 to 60 breaths per minute)
• Restlessness, pallor, or cyanosis
• Hypoxia recorded by either blood gas (partial pressure of oxygen [PO2] less than 60 mm Hg) or pulse oximetry (less than 92% on room air)
• Rising partial pressure of carbon dioxide (PCO2) (recorded by blood gas)
• Inability to tolerate oral feedings
• Depressed sensorium
820
• Presence of chronic cardiovascular or immunodeficiency disease
• Parent unable to manage at home for any reason
In-hospital management focuses on supportive care, focusing on suctioning of nares, humidified supplemental oxygen, and elevation of
the child to a sitting position at a 30- to 40-degree angle. IV hydration (or in infants nasogastric hydration) is needed when respiratory distress
interferes with nursing or bottle feeding.
Occasionally a hospitalized child is not able to be quickly weaned back to room air. Home management of these infants requiring oxygen
is sometimes difficult and may require a team approach, including involvement of a pediatric health care provider and home care nursing
visits. Strict outpatient follow-up is mandatory for as long as the child is receiving home oxygen.
Complications
The first 48 to 72 hours after the onset of cough are the most critical. Apneic spells are common in infants. The child is ill-appearing and
toxic but gradually improves. The fatality rate associated with bronchiolitis is about 1% to 2%. Infants younger than 12 weeks old and those
with underlying cardiorespiratory or immunodeficiency are at risk for severe disease.
Prolonged apnea, uncompensated respiratory acidosis, and profound dehydration secondary to loss of water from tachypnea and an
inability to drink are the factors leading to death in young infants with bronchiolitis. In some children, bronchiolitis can cause minor
pulmonary function problems and a tendency for bronchial hyperreactivity that lasts for years. RSV bronchiolitis has been associated with the
development of asthma, but its role in the causality of asthma is still debated. Recurrent episodes of wheezing can be seen during childhood
in patients with a history of bronchiolitis. This persists into adolescence with 10% of the children still wheezing. However, this figure may
not be different from the general population (Welliver, 2009).Prevention
Palivizumab (Synagis) is an RSV-specific monoclonal antibody used to provide some protection from severe RSV infection for high-risk
infants (see Chapter 24 for guidelines). Educate caregivers about decreasing exposure to and transmission of RSV, especially those with
high-risk infants. Advice should include limiting exposure to child care centers whenever possible; use of alcohol-based hand sanitizers if
available or hand washing if the alcohol-based hand sanitizer is not available (Ralston et al, 2014); avoiding tobacco smoke exposure; and
scheduling RSV prophylaxis vaccination, when indicated.
Asthma
Asthma is a chronic respiratory disease characterized by periods of coughing, wheezing, respiratory distress, and bronchospasm. Asthma can
occur with a persistent cough without significant wheezing. It is the most common chronic respiratory disease of children, with an incidence
as high as 30% of children in the Western world, and it is the leading cause of emergency department visits (Jackson et al, 2011; Liu et al,
2011).
The pathophysiology is the result of immunohistopathologic responses that produce shedding of airway epithelium and collagen
deposition beneath the basement membrane, edema, mast cell activation and inflammatory infiltration by eosinophils, lymphocytes (Th2-like
cells), and neutrophils (especially in fatal asthma). The persistent inflammation can result in irreversible changes, such as airway wall
remodeling. Inflammation causes acute bronchoconstriction, airway edema, and mucous plug formation. In addition, airway inflammation
can trigger a hyperresponsiveness to a variety of stimuli, including allergens, exercise, cold air, and physical, chemical, or pharmacologic
agents. This results in bronchospasm, which presents as wheezing, breathlessness, chest tightness, and cough that can be worse at night or
with exercise. The airflow obstruction is often reversible, either spontaneously or with treatment. Remodeling of the airway can occur
secondary to persistent fibrotic changes in the airway lining. The fibrosis alters the airway caliber, leading to decreased airflow with
permanent changes starting in childhood, but become recognizable in adults. Recent advances have shown that there are different
―phenotypes‖ of this disease with different clinical manifestations, and data suggest that children who have symptoms before 3 years old are
more likely to have changes in lung functioning at 6 years old (Szefler et al, 2014).
Asthma in children is classified as intermittent, mild persistent, moderate persistent, or severe persistent depending on symptoms,
recurrences, need for specific medications, and pulmonary function measurements (Table 25-2). Children classified at any level of asthma
can have episodes involving mild, moderate, or severe exacerbations. Exacerbations involve progressive worsening of shortness of breath,
cough, wheezing, chest tightness, or any combination of these symptoms. The degree of airway hyperresponsiveness is usually related to the
severity of asthma that can change over time. A well-controlled child with asthma has only one exacerbation in 3 years on average (Jackson
et al, 2011).
TABLE 25-2
Classification of Asthma Severity in Children: Clinical Features Before Treatment
Classification and Step Symptoms* Nighttime Symptoms Lung Function
Step 1: Intermittent
Symptoms two times or less per week
Asymptomatic and normal PEF between exacerbations
Requires SABA 2 days/week
Exacerbations brief (few hours or days); varying intensity
No interference with normal activity
Two times or less per month FEV1 >80% predicted
Normal FEV1 between
exacerbations
Step 2: Mild persistent
Symptoms more than two times per week but less than
one time per day
Requires SABA more than two days/week but not more
than one per day
Exacerbations may affect activity (minor)
Three to four times per month FEV1 >80% predicted
Step 3: Moderate
persistent
Daily symptoms
Daily use of inhaled SABA
Some limitations
Exacerbations affect activity, two times or more per
week; may last days
More than one time per week but
not nightly
FEV1 >60% but <80% predicted
Step 4: Severe
persistent
Continual symptoms
Requires SABA several times/day
Extremely limited physical activity
Frequent exacerbations
Often seven times per week FEV1 <60% predicted
*Having at least one symptom in a particular step places the child in that particular classification.
FEV1, Forced expiratory volume in 1 second; PEF, peak expiratory flow; SABA, short-acting beta2-agonist.Adapted from National Heart, Lung, and Blood Institute (NHLBI): Full report of the expert panel: guidelines for the diagnosis and management of asthma,
(EPR-3), Bethesda, MD, 2007, National Institutes of Health.
Many children experience early- and late-phase responses to their asthma episode. The early asthmatic response (EAR) phase is
characterized by activation of mast cells and their mediators, with bronchoconstriction being the key feature. EAR starts within 15 to 30
minutes of mast cell activation and resolves within approximately 1 hour if the individual is removed from the offending allergen. The latephase asthmatic response is a prolonged inflammatory state that usually follows the EAR within 4 to 12 hours after exposure to the allergen,
is often associated with airway hyperresponsiveness more severe than the EAR presentation, and can last from hours to several weeks.
Exercise-induced bronchospasm describes the phenomenon of airway narrowing during, or minutes after, the onset of vigorous activity. Most
asthmatics exhibit airway hyperirritability after vigorous activity and display exercise-induced bronchospasm. For some children, exercise is
the trigger for their asthma. Although asthma is not always associated with an allergic disorder in children, many pediatric patients with
chronic asthma have an allergic component. Increased weight gain in pregnancy and the first 2 years of life may increase TNF-α, a
proinflammatory cytokine implicated in asthma, which may be a predictive biomarker for asthma (Szefler et al, 2014).
It is not known for certain whether hyperresponsiveness of the airways is present at birth or acquired later in genetically predisposed
children. However, the genetic predisposition for the development of an IgE-mediated response to common aeroallergens, known
as atopy,remains the strongest identifiable predisposing risk factor for asthma. A combination of genetic predisposition and exposure to
certain environmental factors are the necessary components responsible for the pathophysiologic response associated with asthma. Origins of
asthma exacerbations include exposure to respiratory virus, seasonal patterns, exposure to mycoplasma pneumonia and Chlamydophila
pneumoniae,pollution, smoking, pregnancy, and psychological stress (Jackson et al, 2011; Szefler, 2013). Asthma is rarely diagnosed before
12 months old due to the high rate of viral illness causing bronchiolitis (Nelson and Zorc, 2013). A diagnosis of asthma should be made with
caution in a toddler who has only wheezing associated with viral infections (Mueller et al, 2013).
The morbidity and mortality statistics of asthma in childhood demonstrate an alarming increasing incidence of asthma and its
complications with a lifetime prevalence of 13% (Nelson and Zorc, 2013). The prevalence rate for asthma is highest among children 5 to 17
years with the 566highest rate among black children (Centers for Disease Control and Prevention, 2015). Minority children have fewer
ambulatory care visits for asthma and are less likely to be on a controller medication. Occupational or environmental exposure can cause
airway inflammation associated with asthma. Factors known to precipitate or aggravate asthma in children include the following:
• Atopic individual response to allergens—inhaled, topical, ingested
• Viral infections and bacterial infections with atypical mycobacterium
• Exposure to known irritants (paint fumes, smoke, air pollutants) and occupational chemicals
• Gastroesophageal reflux
• Exposure to tobacco smoke (for infants, especially smoking by mother)
• Environmental changes—rapid changes in barometric pressure, temperature, especially cold air
• Exercise and psychological factors or emotional stresses (e.g., crying, laughter, anxiety attack, or panic or panic disorder)
• AR and sinusitis
• Drugs (e.g., acetaminophen, aspirin, beta-blockers)
• Food additives (sulfites)
• Endocrine factors (e.g., obesity)
Allergen-induced asthma results in hyperresponsive airways. The majority of children with asthma show evidence of sensitization to any
of the following inhalant allergens:
• House dust mites, cockroaches, indoor molds
• Saliva and dander of cats and dogs
• Outdoor seasonal molds
• Airborne pollens—trees, grasses, and weeds
• Food allergy, including egg and tree nut
Clinical Findings
History
In a primary care setting, asthma should be monitored using a standardized instrument, which may include the Asthma Control Test (ACT),
Asthma Control Questionnaire, Asthma Therapy Assessment Questionnaire, Asthma Control Score, and other instruments as found in the
guidelines summary (National Heart, Lung, and Blood Institute [NHLBI], 2007, p 17). The advantages of a standardized questionnaire are
that it allows the health care provider to assess changes in the patient's asthma and alter the management plan as needed. However, data
suggest the use of these tools is not effective in poorly controlled children in an acute setting (Szefler, 2014).
The assessment of asthma symptoms allows providers to determine if the asthma is well controlled, less well controlled, or poorly
controlled (Mueller et al, 2013). Well-controlled children have symptoms less than 2 days a week and use short-acting beta2-agonists
(SABAs) less than twice 567a week, whereas less well-controlled patients have symptoms more than 2 days a week and likely need a step up
in treatments. Poorly controlled children have symptoms during the day and may utilize SABAs several times a day.
In primary care settings and the emergency department, the initial presentation is assessed based on the ability to talk in sentences,
breathlessness, and alertness (Nelson and Zorc, 2013). Critical points to cover in the history of a child being seen for asthma include the
following:
• Family history of asthma or other related allergic disorders (e.g., eczema or AR)• Conditions associated with asthma (e.g., chronic sinusitis, nasal polyposis, gastroesophageal reflux, and chronic otitis media)
• Complaints of chest tightness or dyspnea
• Cough and wheezing particularly at night and in the early morning or shortness of breath with exercise or exertion (characteristic of asthma)
• Seasonal, continuous, or episodic pattern of symptoms that may be associated with certain allergens or triggering agents
• Episodes of recurrent ―bronchitis‖ or pneumonia
• Precipitation of symptoms by known aggravating factors (upper respiratory infections, acetaminophen, aspirin)
• Level of alertness
Physical Examination
Table 25-3 outlines the physical assessment findings correlated with asthma severity. Broadly speaking, the following may be
seen on physical examination:
• Heterophonous wheezing (different pitches but may be absent if severe obstruction)
• Continuous and persistent coughing
• Prolonged expiratory phase, high-pitched rhonchi especially at the bases
• Diminished breath sounds
• Signs of respiratory distress, including tachypnea, retractions, nasal flaring, use of accessory muscles, increasing restlessness,
apprehension, agitation, drowsiness to coma
• Tachycardia, hypertension or hypotension, pulsus paradoxus
• Cyanosis of lips and nail beds if hypoxic
• Possible associated findings include sinusitis, AD, and AR.
Physical Assessment of Asthma and Asthma Severity
Severity of Asthma Physical Assessment Findings
Mild
Wheezing at the end of expiration or no wheezing
No or minimal intercostal retractions along posterior axillary line
Slight prolongation of expiratory phase
Normal aeration in all lung fields
Can talk in sentences
Moderate
Wheezing throughout expiration
Intercostal retractions
Prolonged expiratory phase
Decreased breath sounds at the base
Severe
Use of accessory muscles plus lower rib and suprasternal retractions; nasal flaring
Inspiratory and expiratory wheezing or no wheezing heard with poor air exchange
Suprasternal retractions with abdominal breathing
Decreased breath sounds throughout base
Impending respiratory arrest
Diminished breath sounds over entire lung filed
Tiring, inability to maintain respirations
Severely prolonged expiration if breath sounds are heard
Drowsy, confused
Diagnostic Studies
Laboratory and radiographic tests should be individualized and based on symptoms, severity or chronology of the disease, response to
therapy, and age. Tests to consider include the following:
• Oxygen saturation by pulse oximetry to assess severity of acute exacerbation. This should be a routine part of every
assessment of a child with asthma. Pulse oximetry measures the oxygen saturation (SaO2) of hemoglobin—the percentage of
total hemoglobin that is oxygenated.
• A CBC if secondary infection or anemia is suspected (also check for elevated numbers of eosinophils).
• Routine chest radiographs are not indicated in most children with asthma. Results are typically normal or only show
hyperinflation. Again imaging should be ordered judiciously with consideration of the long-term risk. However, chest
radiographs can be useful in the following situations: selected cases of asthma or suspected asthma or if the child haspersistent wheezing without a clinical explanation. Children with hypoxia, fever, suspected pneumonia, and/or localized rales
requiring admission are candidates for imaging. Infants with wheezing during the winter who have clinical bronchiolitis do
not need imaging (Nelson and Zorc, 2013).
• If sinusitis is suspected as the trigger, no diagnostic radiographic testing is needed.
• Allergy evaluation should be considered, keeping in mind that history and physical examination are key in this consideration.
(Refer child to pediatric allergist.)
• Sweat test should be considered based on history in every patient with asthma.
• Pulmonary function tests:
• Spirometry testing is the gold standard for diagnosing asthma and should be used on a regular basis to monitor, evaluate,
and manage asthma. Exercise challenges using spirometry can also be done to evaluate the child with exercised-induced
asthma. Children older than 5 years can typically perform spirometry.
568
• To evaluate the accuracy of the spirometry, look for an initial sharp peak with an extension down to the baseline at the
end of expiration that is reproducible at least two times. Compare the child's values with the predicted value for the
child's age, height, sex, and race.
• Look at the forced expiratory volume in 1 second (FEV1), which represents the amount of air exhaled in 1 second. The
interpretation of percentage predicted is:
• >75%: Normal
• 60% to 75%: Mild obstruction
• 50% to 59%: Moderate obstruction
• <49%: Severe obstruction
• The forced vital capacity (FVC) represents the amount of air expelled:
• 80% to 120%: Normal
• 70% to 79%: Mild reduction
• 50% to 69%: Moderate reduction
• <50%: Severe reduction
• The FEV1/FVC represents the amount of air expelled in the first second over the total amount of air expelled and should be
greater than 90% of the predicted value. Spirometry testing is done prior to a breathing treatment and 10 minutes after
the treatment. If the child's FEV1 improves by 12%, the child likely has asthma because this illustrates
hyperresponsiveness.
• The forced expiratory flow (FEF) (FEF25 to FEF75) reflects the middle portion of the downward limb of the curve and is a
good measure of smaller airway function. The interpretation of percentage predicted is:
• >60%: Normal
• 40% to 60%: Mild obstruction
• 20% to 40%: Moderate obstruction
• <10%: Severe obstruction
• Doing spirometry during well-child checks and for sick visits gives the practitioner an excellent indication of the amount
of inflammation and bronchospasm present in the airway (Kamakshya, 2012). Table 25-4 represents abnormal spirometry
patterns.
TABLE 25-4
Abnormal Spirometry Findings
Obstructive Restrictive
FVC Normal or ↓ ↓
FEV1 ↓ ↓Obstructive Restrictive
FEV1/FVC ↓ Normal or ↑
FEV1, Forced expiratory volume in 1 second; FVC, forced vital capacity.
• Consider the use of more sophisticated pulmonary laboratory studies for the child with severe asthma.
• Peak flow measurements:
• If spirometry is not an option, peak expiratory flow (PEF) can be used.
• PEF can be used in some children as young as 4 to 5 years old. The values are instrument specific, so the child's personal
best value is the best guide to help detect possible changes in airway obstruction. The predicted range for height and age
can be substituted if personal best rate is not available (Table 25-5). Interpretation of PEF reading is as follows if PEF is in
the:
• Green zone: More than 80% to 100% of personal best signals good control.
• Yellow zone: Between 50% and 79% of personal best signals a caution.
569
• Red zone: Between 0% and 50% of personal best signals major airflow obstruction.
• Box 25-3 describes use of peak flowmeter and interpretation of results.
Exhaled nitric oxide (Dweik et al, 2011):
• A biomarker for the children with asthma is exhaled nitric oxide testing, which measures a fraction of exhaled nitric oxide (FEno).
• The test measures eosinophilic airway inflammation and helps to determine whether corticosteroids would be helpful in the
management of the patient. It may support the diagnosis of asthma and can help determine compliance with corticosteroid therapy.
• A FEno value of more than 35 ppb in children indicates eosinophilic inflammation and likely responsiveness to corticosteroids, whereas
25 to 35 ppb should be interpreted with caution. There is still controversy about this test, although guidelines have been published.
Management
Management strategies are based on whether the child has intermittent, mild persistent, moderate persistent, or severe persistent asthma
(see Table 25-3). A stepwise approach is recommended. If control of symptoms is not maintained at a particular step of classification and
management, the health care provider first should reevaluate for adherence and administration factors. If these factors do not appear to be
responsible for the lack of symptom control, go to the next treatment step. Likewise, gradual step-downs in pharmacologic therapy may be
considered when the child is well controlled for 3 months. Inhaled corticosteroids may be reduced about 25% to 50% every 3 months to the
lowest possible dose needed to control the child's asthma (NHLBI, 2007; Szefler et al, 2014).
Chronic Asthma
Treatment of chronic asthma in children is based on general control measures and pharmacotherapy. Control measures can
include the following:
• Avoid exposure to known allergens or irritants.
• Avoid use of acetaminophen in children at risk for asthma (Jackson et al, 2011; McBride, 2011).
• Administer yearly influenza vaccine.
• Control environment to eliminate or reduce offending allergen.
• Consider allergen immunotherapy. Studies have pointed to reduction in health care cost and improved outcomes associated
with allergy immunotherapy (Dretzke et al, 2013; Hankin et al, 2013).
• Treat rhinitis, sinusitis, or gastroesophageal reflux.
• Other pharmacologic agents that may need to be considered include:
• Anticholinergics—to reduce vagal tone in the airways (may also decrease mucus gland secretion)
• Cromolyn sodium—to inhibit mast cell release of histamine
• Leukotriene modifiers—to disrupt the synthesis or function of leukotrienes
• If needed, refer to pulmonology for omalizumab, a recombinant DNA-derived, humanized IgG monoclonal antibody that
binds to human IgE on the 570surface of mast cells and basophils. This anti-IgE monoclonal antibody is used as a secondline treatment for children older than 12 who have moderate to severe allergy-related asthma and react to perennial
allergens. It is used when symptoms are not controlled by inhaled corticosteroids.• Follow up with PCP after an exacerbation requiring emergency department care, and obtain a clear written asthma action
plan.
• Education regarding asthma basics, including triggers and prevention with environmental modification, as well as the
different treatment modalities includes the techniques of administration and dispelling any myths regarding asthma
medication. In terms of coping, the child and family need to be able to understand their emotions, worries, and uncertainty, as
well as when to contact their PCP. Developing and understanding the asthma action plan is very important during a well-child
visit (Archibald and Scott, 2014).
The pharmacologic management of asthma in children is based on the severity of asthma and the child's age. The stepwise
approach to treatment (Figs. 25-1 and 25-2) is based on severity of symptoms and the use of pharmacotherapy to control chronic
symptoms, maintain normal activity, prevent recurrent exacerbations, and minimize adverse side effects and nearly “normal”
pulmonary function. Within any classification, a child may experience mild, moderate, or severe exacerbations. NHLBI
guidelines for assessing asthma control and initiating and adjusting asthma therapy for the various pediatric age groups are
found in Figures 25-3 and 25-4.
Important considerations to note in the pharmacologic treatment of asthma include the following:
• Control of asthma should be gained as quickly as possible by starting at the classification step most appropriate to the initial
severity of the child's symptoms or at a higher level (e.g., a course of systemic corticosteroids or higher dose of inhaled
corticosteroid). After control of symptoms, decrease treatment to the least amount of medication needed to maintain control.
• Systemic corticosteroids may be needed at any time and stepped up if there is a major flare-up of symptoms. 573Control of
inflammation is a key principle in the management of asthma.
• The combination of inhaled corticosteroids with a long-acting beta2-agonist (LABA) can further control asthma (Szefler, 2013).
• Children with intermittent asthma may have long periods in which they are symptom-free; they can also have life-threatening
exacerbations, often provoked by respiratory infection. In these situations, a short course of systemic corticosteroids should be
used.
• Variations in asthma necessitate individualized treatment plans.
• β2 agonists can be administered with metered dose inhaler (MDI) therapy via spacer for children with mild and moderate
exacerbations of asthma, but for children with severe airway obstruction who may have decreased deposition of drug in the
base of the lung, a nebulizer may be better (Nelson and Zorc, 2013). There is need for more research on the use of MDI therapy
and nebulizer therapy in the pediatric population (Szefler et al, 2014). A spacer or holding chamber with an attached mask
enhances the delivery of MDI medications to the lower airways of a child. Spacers eliminate the need to synchronize inhalation
with activation of MDI. Older children can use a spacer without the mask.
• Dry powder inhalers (DPIs) do not need spacers or shaking before use. Instruct children to rinse their mouth with water and
spit after inhalation. DPIs should not be used in children younger than 4 years old.
• Different inhaled corticosteroids are not equal in potency to each other on a per puff or microgram basis. Tables 25-6 and 25-
7 compare daily low, medium, and high doses of various inhaled corticosteroids used for children. Combination inhaled
corticosteroid and LABA can be used in children from 4 years old (Taketomo et al, 2014).
For treatment of exercise-induced bronchospasm:
• Warm up before exercise for 5 to 10 minutes.
• Use either an inhaled SABA or a mast cell stabilizer (cromolyn) or both prior to exercise. Combination of both types of
drugs is the more effective therapy. A LABA can be used in older children.
• Use two puffs of a β2 agonist and/or cromolyn MDI 15 to 30 minutes before exercise. Tolerance may develop if a β2 agonist
is used more than a few times 574a week; it should not be used as a controller monotherapy. Those who exercise
regularly and develop symptoms of asthma should use controller medication, preferably an inhaled corticosteroid.
• Using a scarf or mask around the mouth may decrease exercise-induced asthma (EIA) induced by cold.
Table 25-8 identifies the usual dosages for long-term control medications (exclusive of inhaled corticosteroids) used to treat
asthma in children. Quick-relief medications are listed in Table 25-9. Practice parameters are guides and should not replace
individualized treatment based on clinical judgment and unique differences among children.
Acute Exacerbations of Asthma
The treatment of acute episodes of asthma is also based on classification of the severity of the episode. Acute episodes 579are
classified as mild, moderate, and severe. Signs and symptoms are summarized in Table 25-10. Early recognition of warning
signs and treatment should be stressed in both patient or parent education, or both.The initial pharmacologic treatment for acute asthma exacerbations is shown in Figure 25-5. It consists of inhaled SABAs
(albuterol), two to six puffs every 20 minutes for three treatments by way of MDI with a spacer, or a single nebulizer treatment
(0.15 mg/kg; minimum 1.25 to 2.5 mg of 0.5% solution of albuterol in 2 to 3 mL of normal saline).
If the initial treatment results in a good response (PEF/FEV1 > 70% of the patient's best), the inhaled SABAs can be continued every 3 to 4
hours for 24 to 48 hours with a 3-day course of oral steroids at 1 to 2 mg/kg/day to a maximum of 60 mg per day. Reassessment is important
to ensure an adequate response and to further assess asthma severity.
An incomplete response (PEF or FEV1 between 40% and 69% of personal best or symptoms recur within 4 hours of therapy) is treated by
continuing β2 agonists and adding an oral corticosteroid. The β2 agonist can be given by nebulizer or MDI with spacer. Parents should be
taught to call their PCP for additional instructions. If there is marked distress (severe acute symptoms) or a poor response (PEF or
FEV1 <40%) to treatment, the child should have the β2 agonist repeated immediately and should be taken to the emergency department.
Emergency medical rescue (911) transportation should be used if the distress is severe and the child is agitated and unable to talk. If children
experience acute asthma exacerbations more than once every 4 to 6 weeks, their treatment plan should be reevaluated.
581
This chapter focuses on the outpatient management of patient with asthma. However, familiarity with other drug options used in more
severe asthma is important. They include:
• Magnesium sulfate IV is used in emergency settings to decrease the intracellular calcium concentration. It causes bronchodilation due to
respiratory smooth muscle relaxation.
• Ipratropium oral inhalation is an anticholinergic bronchodilator used to treat bronchospasms. Evidence of its long-term maintenance use to
control bronchospasms is lacking (Taketomo et al, 2014)
• Epinephrine given subcutaneously or intramuscularly is still an option in severe asthma where the delivery of medication to smaller airways
is limited due to bronchoconstriction.
• Heliox is a mixture of oxygen and helium, which can improve drug delivery in obstructed airways because helium has a lower density and
less airway resistance (Nelson and Zorc, 2013).
Complications
Complications from asthma can range from mild secondary respiratory infections to respiratory arrest. Unresponsiveness to pharmacologic
agents can lead to status asthmaticus and ultimately to death. Chronic high-dose steroid use leads to growth retardation and other related side
effects.
Patient and Parent Education and Prevention
The PCP needs to support self-care management through in-depth education as appropriate. Easy to understand education needs to be tailored
to meet the child's individual 582needs, family needs, and cultural beliefs using a ―teach back‖ technique. Correct administration of inhaled
medication should be demonstrated during initial training sessions and reevaluated in subsequent visits. Provide instruction on the following:
• Basic understanding of what asthma is, what is good asthma control, and what is the child's current level of symptomatology
• Environmental control of allergens or triggers, such as smoking and dust
• Basic understanding of what different medications do and how to use them: Give clear, written instructions on how to
administer, how much and when to give, monitoring side effects, and how long medication should be taken. A written plan is
highly recommended based on either symptoms or peak expiratory flow rate (PEFR).
• How to use inhalers, spacer devices, or aerosol equipment (Box 25-4) along with proper cleaning of aerosol equipment
Milia
Milia are multiple, firm, pearly, opalescent white papules scattered over the forehead, nose, and cheeks. Their intraoral
counterparts are called Epstein pearls. Histologically, milia represent superficial epidermal inclusion cysts filled with keratinous
material associated with the developing pilosebaceous follicle. No treatment is necessary because milia exfoliate spontaneously
in most infants over the first few weeks of life (Fig. 39-6).
Vascular and Pigmented Nevi
Nevi are a common finding in children. The two most common types are vascular nevi (vascular malformations and
hemangiomas) and pigmented nevi (e.g., mongolian spots, café au lait spots, acquired melanocytic nevi, atypical nevi, and
lentigines).
Vascular nevi are caused by a structural abnormality (malformations) or by an overgrowth of blood vessels (hemangiomas)
and are flat, raised, or cavernous. Flat lesions or vascular malformations include salmon patches (also called macular stains), an
innocent malformation that is a light red macule appearing on the nape of the neck, upper eyelids, and glabella. Approximately
60% to 70% of newborns have a salmon patch on the back of the neck. Port-wine stains occur in 0.2% to 0.3% of newborns
(Cohen, 2013). At 1 year old, 10% to 12% of Caucasian infants have a hemangioma—females three times more likely than males.
There is also an increased incidence of hemangioma in premature neonates. Vascular malformations are always present at birthand do not resolve spontaneously. Precursor lesions of hemangiomas are present at birth 50% of the time. They undergo rapid
growth (proliferative stage), stability (plateau phase), and regression (involution phase); 90% are completely resolved in
children 9 to 10 years old (Paller and Mancini, 2011).
Pigmented nevi are caused by an overgrowth of pigment cells. Pigmented nevi most commonly seen are mongolian spots
(found in up to 90% of African Americans, 62% to 86% of Asians, 70% of Hispanics, and less than 10% of Caucasians), café au
lait spots (found in up to 33% of normal children and in 50% of patients with McCune-Albright syndrome), and acquired
melanocytic nevi, the most common tumor of childhood. Atypical nevi, also called dysplastic nevi, are potential precursors for
malignant melanoma. Dysplastic nevi are uncommon under 18 years old but have a higher incidence in melanoma-prone
families (Paller and Mancini, 2011).
Clinical Findings
History
• Presence from birth, or age first noted
• Progression of lesion
• Familial tendencies for similar nevi, especially for history of melanoma
1031
Physical Examination
Findings include the following (Box 37-8):
• Vascular malformations or flat vascular nevi are present at birth and grow commensurate with the child's growth.
• Hemangiomas are classified as superficial, deep (cavernous), or mixed. They may or may not be present at birth, but they usually emerge by
2 to 3 weeks of life. They may manifest initially as a pale macule, a telangiectatic lesion, or a bright red nodular papule. After appearing,
hemangiomas go through a proliferative phase during which they grow rapidly and form nodular compressible masses, ranging in size from
a few millimeters to several centimeters. Occasionally they may cover an entire limb, resulting in asymmetric limb growth. Rapidly
growing lesions may ulcerate. The final phase of involution occurs slowly (10% per year) but spontaneously (30% by 3 years old, 50% by 5
years old, 70% by 7 years old, and 90% by 9 to 10 years old). Average involution begins between 12 and 24 months old, heralded by gray
areas in the lesion followed by flattening from the center outward. Most hemangiomas appear as normal skin after involution, but others
may have residual changes, such as telangiectasias, atrophy, fibrofatty residue, and scarring (Paller and Mancini, 2011).
• Pigmented nevi may be present at birth or acquired during childhood.
• Atypical nevi are larger than acquired nevi; have irregular, poorly defined borders; and have variable pigmentation.
Box 37-8
C o mmo n Va s cula r and Pig men t ed L es ion s
I. Vascular malformations or flat vascular nevi
A. Salmon patch or nevus flammeus: Light pink macule of varying size and configuration. Commonly seen on the glabella, back of neck,
forehead, or upper eyelids.
B. Port-wine stain: Purple-red macules that occur unilaterally and tend to be large. Usually occur on face, occiput, or neck, although they
may be on extremities.
II. Hemangiomas
A. Superficial (strawberry) hemangiomas are found in the upper dermis of the skin and account for the majority of hemangiomas.
B. Deep cavernous hemangiomas are found in the subcutaneous and hypodermal layers of the skin; although similar to superficial
hemangiomas, there is a blue tinge to their appearance. With pressure, there is blanching and a feeling of a soft, compressible tumor.
Variable in size, they can occur in places other than skin.
C. Mixed hemangiomas have attributes of both superficial and deep hemangiomas.
III. Pigmented nevi
A. Mongolian spots: Blue or slate-gray, irregular, variably sized macules. Common in the presacral or lumbosacral area of dark-skinned
infants; also on the upper back, shoulders, and extremities. The majority of the pigment fades as the child gets older and the skin
darkens. Solitary or multiple, often covering a large area.
B. Café au lait spots: Tan to light brown macules found anywhere on the skin; oval or irregular shape; increase in number with age.
C. Acquired melanocytic nevi are benign, light brown to dark brown to black, flat, or slightly raised, occurring anywhere on the body,
especially on sun-exposed areas above the waist.
1. Junctional nevi represent the initial stage, with tiny, hairless, light brown to black macules.
2. Compound nevi—a few junctional nevi progress to more elevated, warty, or smooth lesions with hair.
3. Dermal nevi are the adult form, dome shaped with coarse hair.
4. Atypical nevi usually appear at puberty, have irregular borders, variegated pigmentation, are larger than normal nevi (6 to
15 mm); usually found on trunk, feet, scalp, and buttocks.5. Halo nevi appear in late childhood with an area of depigmentation around a pigmented nevus, usually on trunk (see Fig. 37-
34).
D. Acanthosis nigricans is velvety brown rows of hyperpigmentation in irregular folds of skin, usually the neck and axilla; tags may also
be present.
E. Lentigines are small brown to black macules 1 to 2 mm in size appearing anywhere on the body in school-age children.
F. Freckles: 1 to 5 mm light brown, pigmented macules in sun-exposed areas.
Differential Diagnosis
Hematomas or ecchymoses of child abuse are occasionally confused with some nevi. Non–insulin-dependent diabetes mellitus (NIDDM)
often causes acanthosis nigricans.
Management
1. Flat vascular nevi
• Salmon patches: Fade with time, usually by 5 or 6 years old; no treatment is needed.
• Port-wine stains: A permanent defect that grows with the child, so cosmetic covering is often used. If forehead and eyelids are
involved, there is potential for multiple syndromes, including Sturge-Weber, Klippel-Trenaunay-Weber, and Parkes Weber.
Neurodevelopmental and ophthalmologic follow-up is needed. Referral to a dermatologist for possible laser treatment or cosmesis
is required.
2. Hemangiomas
• Reassure and educate the family about the nature and course of these nevi and that they are not related to anything the mother did
during pregnancy.
• Follow-up frequently, especially during the proliferation phase. Sequential photographs are helpful.
• If the lesions are strategically placed (eye, lip, oral cavity, ear, airway, diaper area), ulcerating, multiple, very large, or grow very
quickly, prompt referral to a dermatologist is indicated because early treatment is most effective. Steroids (intralesional and
oral) are prescribed during the proliferation phase until growth is stabilized, then gradually tapered. Indications for steroid
treatment are interference with physiologic functions (e.g., breathing, hearing, eating, and vision), recurrent bleeding or ulceration,
high-output 1032congestive heart failure, Kasabach-Merritt syndrome, rapid growth that distorts facial features, or presence in the
diaper area. Interferon-alpha may also be used. Treatment by surgery, cryotherapy, radiation, or injecting sclerosing agents often
leads to scarring. Large, deep lesions can cause cardiovascular complications, disseminated intravascular coagulation, or
compression of internal organs.
• Involution (without treatment) occurs at a rate of 10% per year. Scarring may be present if ulceration occurs; fibrofatty masses,
atrophy, and telangiectasis can occur following involution. Laser therapy is effective management for residual telangiectasias
(Paller and Mancini, 2011).
3. Pigmented nevi: Educate family about the nature of these lesions.
• Mongolian spots: Document to distinguish from bruise; fade with time, usually no traces by adulthood.
• Blue nevus: Heavily pigmented melanocytes in papule or nodule that can develop melanoma.
• Café au lait spots: If six or more lesions larger than 5 mm in diameter are present in children younger than 15 years old and more
than 1.5 cm in diameter for older individuals, or if axillary freckling (Crowe's sign), neurofibromas, or iris hamartomas is also
present, refer child to rule out neurofibromatosis, McCune-Albright syndrome, tuberous sclerosis, LEOPARD* syndrome,
epidermal nevus syndrome, Bloom syndrome, ataxia-telangiectasia, and Silver-Russell syndrome (Cohen, 2013).
Complications
Ulceration, infection, platelet trapping, airway or visual obstruction, or cardiac decompensation can occur with large vascular nevi.
Kasabach-Merritt syndrome occurs when thrombocytopenic hemorrhage occurs in a large, deep hemangioma. Melanoma in congenital nevi is
possible. An autosomal dominant, familial, atypical mole and melanoma syndrome has been identified genetically. Children with multiple
atypical nevi and family members with melanoma are at risk for childhood melanoma.
Patient and Family Education
Monitoring nevi that are at risk for developing melanoma is important as is teaching the family to watch nevi for any changes. Changes of
particular concern are development of an off-center nodule or papule, color change, bleeding, persistent irritation, erosion, ulceration, and
rapid growth. See Chapter 39 for more information.
1033
Impetigo
Impetigo is a common contagious bacterial infection of the superficial layers of the skin. It has two forms: nonbullous, with
honey-colored crusts on the lesions, and bullous (Fig. 37-4). Impetigo is usually caused by group A streptococcus (Streptococcus
pyogenes), Staphylococcus aureus,or MRSA. Often streptococcus and staphylococcus can be 992cultured from an impetigo lesion.
Nonbullous impetigo accounts for more than 70% of cases, with S. aureus as the most common pathogen. Nonbullous impetigo
usually follows some type of skin trauma (e.g., bites, abrasions, or varicella) or another skin disease, such as atopic dermatitis.Bullous impetigo occurs sporadically, develops on intact skin, and is more common in infants and young children. Certain
epidermal types of S. aureus produce a toxin that causes bullous skin lesions.
Bacterial colonization of the skin occurs several days to months before lesions appear; the organism usually spreads from autoinoculation
via hands, towels, clothing, nasal discharge, or droplets. Impetigo occurs more frequently with poor hygiene; during the summer months; in
warm, humid climates; and in lower socioeconomic groups. Streptococci that cause pharyngitis rarely cause impetigo and vice versa.
Secondary bacterial infections of underlying skin problems (dermatitis, varicella, psoriasis) are most commonly caused by staphylococci
(Cohen, 2013).
Clinical Findings
History
• Pruritus, spread of the lesion to surrounding skin, and earlier skin disruption at the site
• Weakness, fever, and diarrhea may accompany bullous impetigo
Physical Examination
The following can be found:
• Nonbullous, classic, or common impetigo—begins as 1- to 2-mm erythematous papules or pustules that progress to vesicles or bullae, which
rupture, leaving moist, honey-colored, crusty lesions on mildly erythematous, eroded skin; less than 2 cm in size; little pain but rapid spread
• Bullous impetigo—large, flaccid, thin-wall, superficial, annular, or oval pustular blisters or bullae that rupture, leaving thin varnish-like
coating or scale
993
• Lesions are most common on face, hands, neck, extremities, or perineum; satellite lesions may be found near the primary site, although they
can be anywhere on the body
• Regional lymphadenopathy
Diagnostic Studies
Gram stain and culture are ordered if identification of the organism is needed in recalcitrant or severe cases.
Differential Diagnosis
Herpes simplex, varicella, nummular eczema, contact dermatitis, tinea, kerion, and scabies are included in the differential diagnosis.
Management
Management involves the following:
• Topical antibiotics may be used if the impetigo is superficial, nonbullous, or localized to a limited area. Topical treatment alone provides
clinical improvement but may prolong the carrier state (Weston and Morelli, 2013). In localized regions, topical antibiotics (such as,
bacitracin, polymyxin B, and neomycin) may be used, but, given the increasing resistance to traditional topical antibiotics, mupirocin and
retapamulin are considered better choices for topical treatment (Cohen, 2013; Weinberg and Tyring, 2010; Weston and Morelli, 2013). Oral
antibiotics are recommended for multiple lesions or nonbullous impetigo with infection in multiple family members, child care groups, or
athletes. Treat for S. aureus and S. pyogenes because coexistence is common (Cohen, 2013).
• Cephalexin: 40 mg/kg/day for 7 to 10 days
• Amoxicillin/clavulanate: 50 to 90 mg/kg/day for 7 to 10 days
• Dicloxacillin: 15 to 50 mg/kg/day for 7 to 10 days
• Cloxacillin: 50 to 100 mg/kg/day for 7 to 10 days
• Clindamycin: 10 to 25 mg/kg/day for 7 to 10 days
• For widespread infection with constitutional symptoms and deeper skin involvement, use an oral antibiotic active against beta-lactamase–
producing strains of S. aureus, such as amoxicillin/clavulanate, dicloxacillin, cloxacillin, or cephalexin.
• If an infant has bullous impetigo, use parenteral beta-lactamase–resistant antistaphylococcal penicillin, such as methicillin, oxacillin, or
nafcillin.
• If there is no response in 7 days, swab beneath the crust, and do Gram stain, culture, and sensitivities. Community-acquired MRSA should
be considered. This organism is more susceptible to clindamycin and trimethoprim-sulfamethoxazole (TMP-SMX) (see Chapter 24 for
treatment of MRSA).
• Educate regarding cleanliness, hand washing, and spread of disease.
• Exclude from day care or school until treated for 24 hours.
• Schedule a follow-up appointment in 48 to 72 hours if not improved.
Complications• Cellulitis may occur with nonbullous impetigo and present in the form of ecthyma (infection involving entire epidermis) or erysipelas
(spreading cellulitis with induration).
• Lymphangitis, suppurative lymphadenitis, guttate psoriasis, erythema multiforme, scarlet fever, or glomerulonephritis may occur following
infection with some strains of Streptococcus. Acute rheumatic fever is a rare complication of streptococcal skin infections.
• Staphylococcal scalded skin syndrome (SSSS) is a blistering disease that results from circulating epidermolytic toxin–producing S.
aureus. SSSS is most common in neonates (Ritter disease), infants, and children younger than 5 years old. It manifests abruptly with
fever, malaise, and tender erythroderma, especially in the neck folds and axillae, rapidly becoming crusty around the eyes, nose, and mouth.
Nikolsky sign (peeling of skin with a light rub to reveal a moist red surface) is a key finding. Treatment may include hospitalization and
parenteral antibiotics, especially for young children (Berk and Bayliss, 2010). Antibiotics of choice are intravenous (IV) or oral
dicloxacillin, a penicillinase-resistant penicillin, first- or second-generation cephalosporins, or clindamycin. Quicker healing without
scarring results if steroids are avoided, there is minimal handling of the skin, and ointments and topical mupirocin are used at the infection
site (Berk and Bayliss, 2010; Patel and Patel, 2010). Severe cases may need treatment similar to extensive burn care.
Patient and Family Education
• Thorough cleansing of any breaks in the skin helps prevent impetigo.
• Postinflammatory pigment changes can last weeks to months.
• The patient should not return to school or day care until 24 hours of antibiotic treatment is completed.
Molluscum Contagiosum
A benign common childhood viral skin infection with little health risk, molluscum contagiosum often disappears on its own in a
few weeks to months and is not easily treated (Fig. 37-18). This poxvirus replicates in host epithelial cells. It attacks skin and
mucous membranes and is spread by direct contact, by fomites, or by autoinoculation (typically scratching). It is commonly
found in children and adolescents. The incubation period is about 2 to 7 weeks but may be as long as 6 months (Weston and
Morelli, 2013). Infectivity is low but the child is contagious as long as lesions are present.
Clinical Findings
History
• Itching at the site
• Possible exposure to molluscum contagiosum
Physical Examination
• Very small, firm, pink to flesh-colored discrete papules 1 to 6 mm in size (occasionally up to 15 mm)
• Papules progressing to become umbilicated (may not be evident) with a cheesy core; keratinous contents may extrude from the umbilication
• Surrounding dermatitis is common
• Face, axillae, antecubital area, trunk, popliteal fossae, crural area, and extremities are the most commonly involved areas; palms, soles, and
scalp are spared
• Single papule to numerous papules; most often numerous clustered papules and linear configurations
• Sexually active or abused children can have genitally grouped lesions
• Children with eczema or immunosuppression can have severe cases; those with human immunodeficiency virus (HIV) infection or AIDS
can have hundreds of lesions
Differential Diagnosis
Warts, closed comedones, small epidermal cysts, blisters, folliculitis, and condyloma acuminatum are included in the differential diagnosis.
Management
• Untreated lesions usually disappear within 6 months to 2 years but may take up to 4 years to completely go away. There is no consensus on
the management of molluscum contagiosum and no evidence-based literature to show that any treatment is superior to placebo. Therapy
may be necessary to alleviate discomfort, reduce itching, minimize autoinoculation, limit transmission, and for cosmetic reasons. Genital
lesions may need to be treated to prevent spread to sexual partners.
• Mechanical removal of the central core is to prevent spread and autoinoculation. Using eutectic mixture of local anesthetics (EMLA) cream
(lidocaine/prilocaine) 30 to 45 minutes before the procedure reduces discomfort. Curettage is done with a sharp blade to remove the papule.
Piercing the papule and expressing the plug is an option but is painful.
• There are reports that irritants (such as, surgical tape, adhesive tape, or duct tape) applied each night can result in lesion resolution.
• Topical medications may prove beneficial. Recheck the patient in 1 to 2 weeks to determine need for retreatment.
• Liquid nitrogen applied for 2 to 3 seconds (easiest but also painful).• Trichloroacetic acid 25% to 50% applied by dropper to the center of the lesion, followed by alcohol (use with caution). Surround the
lesion first with petroleum jelly.
• Cantharidin 0.7% in collodion applied by dropper to the center of the lesion, followed by alcohol. Salicylic or lactic acid or KOH or
podophyllin can also be used.
• Podofilox 0.5% topical solution or gel, or imiquimod 5% applied daily with a toothpick or cotton-tipped swab.
• Tretinoin or tazarotene cream or gel applied to lesion each night.
• Silver nitrate, iodine 7% to 9%, or phenol 1% applied for 2 to 3 seconds.
1008
• Cimetidine 30 to 40 mg/kg/day in two divided doses orally for 6 weeks if topical treatment fails.
• Sexual abuse of children with genitally grouped lesions should be suspected and evaluated.
• Evaluate for HIV infection if hundreds of lesions are found.
• Wait and see approach—spontaneous clearing occurs over years.
Complications
Molluscum dermatitis, a scaly, erythematous, hypersensitive reaction, can occur and will respond to moisturizer; avoid hydrocortisone
because it causes molluscum to flare. Impetiginized lesions, inflammation of the eyes or conjunctiva, and scarring can occur.
Patient and Family Education
Patients are contagious, but there is no need to exclude them from day care or school. Children with impaired immunity, atopic dermatitis, or
traumatized skin are at greater risk for broader spread. Severe inflammation is possible several hours after application of cantharidin. Scarring
is unusual.
Warts
Warts are common childhood skin tumors characterized by a proliferation of the epidermis and mucosa infected by the human papillomavirus
(HPV). There are over 100 HPV types, and each one produces characteristic lesions in specific locations (e.g., verruca vulgaris, verruca
plana, verruca plantaris, and condyloma acuminatum). Trauma promotes inoculation of the HPV (Koebner phenomenon); as a result, most
warts are on the hands, fingers, elbows, and plantar surfaces of the feet.
The transmission of warts from person to person depends on viral and host factors, such as quantity of virus, location of warts, preexisting
skin injury, and cell-mediated immunity. Transmission is from fomites or skin-to-skin contact, and autoinoculation is frequent. Incubation is
from 1 to 6 months, possibly years.
Although a large percentage of all warts resolve spontaneously within 3 to 5 years, there is a high recurrence rate. Cutaneous warts are
rarely a serious health concern but present cosmetic problems for children and their families (Cohen, 2013).
Clinical Findings
History
The history can include exposure to someone with warts. Though most common on the extremities, warts can occur anywhere on the body,
including the face, scalp, and genitalia.
Physical Examination
• Common warts (verruca vulgaris) are usually elevated flesh-colored single papules with scaly, irregular surfaces and
occasionally black pinpoints, which are thrombosed blood vessels. They are usually asymptomatic and multiple and are found
anywhere on the body, although most commonly on the hands, nails, and feet. They may be dome shaped, filiform, or
exophytic (Fig. 37-19). Filiform warts project from the skin on a narrow stalk and are usually seen on the face, lips, nose,
eyelids, or neck. Periungual warts are common, occurring around the cuticles of the fingers or toes.
Plantar warts (verrucae plantaris or mosaic) are commonly found on weight-bearing surfaces of the feet. They grow inward and
disrupt skin markings.
• Flat warts (verruca plana or juvenile warts) are seen commonly on the face, neck, and extremities. They are small, slightly
elevated papules and number from few to several hundred.
• Condylomata acuminata on genital mucosa and adjacent skin are multiple, confluent warts with irregular surfaces, light color,
and cauliflower-like appearance (Fig. 37-20).
Differential Diagnosis
The differential diagnosis includes calluses, corns, foreign bodies, moles, comedones, and squamous cell carcinoma.
ManagementThere is no single effective treatment for warts; watchful waiting is an option. The recurrence rate is high; they typically do not resolve with
just a single treatment. No treatment is necessary if the warts are asymptomatic. The decision to treat should be based on location, number
and size of lesions, discomfort, and whether they are cosmetically objectionable. Treatment should not be harmful, and scarring should be
avoided. Genital warts found in young children or in adolescents who are not sexually active should create suspicion of sexual abuse.
Specific treatment options are outlined in Box 37-6. Follow up in 2 to 3 weeks to evaluate response.
Complications
Scarring from removal can occur. A ring of satellite warts may develop at the edge of the blister following treatment with cantharidin.
Immunocompromised hosts can have extensive involvement.
1009
Patient and Family Education
A blister, sometimes hemorrhagic, may form 1 to 2 days after liquid nitrogen treatment. Redness and itching may herald regression of a wart.
Parents and patients must be warned that multiple or prolonged treatment is often necessary.
Herpetic whitlow, occurring on a finger or thumb, is a swollen, painful lesion with an erythematous base and ulceration
resembling a paronychia. It occurs on fingers of thumb-sucking children with gingivostomatitis or adolescents with genital HSV
infection.
Management
Management can be guided by considering the host (e.g., age, area and extent of involvement, and immune status) and the drug
needed (Table 37-5). Treatment includes:
1. Burow solution compresses three times a day to alleviate discomfort
2. Acyclovir 20 to 40 mg/kg/dose orally five times a day for 5 days, or 200 mg five times a day for 7 to 10 days (maximum
pediatric dose 1000 mg/day) may be indicated to help shorten the course and alleviate symptoms for children older than 2
years old with the following conditions:
• Any underlying skin disorder (e.g., eczema)
• A severe case
• An immunocompromised disease
• Systemic symptoms with primary genital infection
• Occasionally for initial severe gingivostomatitis
Acyclovir is most effective if started within 3 days of disease onset. Famciclovir or valacyclovir are additional antiviral
agents approved for use in adults.
3. Topical acyclovir ointment may help for initial genital herpes infections but is often not beneficial for recurrent infections.
4. Oral acyclovir 200 mg five times a day for 5 to 10 days may speed healing of herpetic whitlow (see Fig. 37-16).
5. Antibiotics for secondary bacterial (usually staphylococcal) infection:
• Mupirocin: Topically three times a day for 5 days
• Erythromycin: 40 mg/kg/day for 10 days
• Dicloxacillin: 12.5 to 50 mg/kg/day for 10 days
6. Oral anesthetics for comfort; use with caution in children (the child needs to be able to rinse and spit):
• Viscous lidocaine 2% topical
• Liquid diphenhydramine alone or combined with aluminum hydroxide or magnesium hydroxide as a 1 : 1 rinse
(maximum of 5 mg/kg/day diphenhydra-mine in case it is swallowed); it can also be applied to the lesions with cottontipped swabs
7. Newborn infant, immunosuppressed child, child with infected atopic dermatitis, or child with a lesion in the eye or on the
eyelid margin; consult with or refer to an appropriate provider
8. Offer supportive care, such as antipyretics, analgesics, hydration, and good oral hygiene
1006
9. Exclude from day care only during the initial course (gingivostomatitis) and if the child cannot control secretions
10. Recurrent, frequent, and severe HSV infection may be treated with acyclovir prophylaxis for 6 months
Patient and Family Education
Recurrence of infection, possible triggering factors, and avoidance measures should be discussed. Triggers can include physical
and psychological stress, trauma, fever, exposure to UV light, illness, menses, and extreme weather. Contagiousness of lesions
and oral secretions must be understood. Explanation of the course of primary disease, with fever lasting up to 4 days and lesions
taking at least 2 weeks to heal, is important.Clostridium
difficile
Unknown
Variety of symptoms
and severity are seen:
mild to explosive
diarrhea, bloody
stools, abdominal
pain, fever, nausea,
vomiting
Mild to moderate illness
is characterized by
watery diarrhea, lowgrade fever, and mild
abdominal pain
During or
after
several
weeks
of
antibiot
ic use;
can
occur
withou
t being
associa
ted
with
such
treatme
nt
Acquired from
the
environment
or from stool
of other
colonized or
infected
people by the
fecal-oral
route
Stool cultures;
enzyme
immunoassa
y for toxin
A, or A and
B; positive
gross blood,
leukocytes;
CBC: ↑
WBCs; ESR
normal
Discontinue current
antibiotic (any antibiotic,
but notably ampicillin,
clindamycin, second- and
third-generation
cephalosporins).
Fluids and electrolyte
replacement are usually
sufficient. If antibiotic is
still needed or illness is
severe, treat with oral
metronidazole (drug of
choice in children) or
vancomycin for 7 to 10
days.
Supplement with
probiotics. Lactobacillus
GG, Saccharomyces
boulardii are
recommended (Jones,
2010; Shane, 2010).
Complications include
pseudomembranous
colitis, toxic megacolon,
colonic perforation,
relapse, intractable
proctitis, death in
debilitated children.
Rotavirus 1 to 3 days;
preval
ent
durin
g
cooler
month
s in
tempe
rate
climat
es
Acute-onset fever,
vomiting, and watery
diarrhea occur 2 to 4
days later in children
<5 years old,
especially those
between 3 to 24
months old
3 to 8 days Fecal-oral; viable
on inanimate
objects;
rarely
contaminate
d water or
food
Enzyme
immunoassa
y and latex
agglutinatio
n assays for
group A
rotavirus
antigen;
virus can be
found by
electron
microscopy
and specific
nucleic acid
amplificatio
n methods.
Supportive care: May need
to correct dehydration
and electrolyte
imbalances. Oral IG has
been used in those
immunocompromised.
Preventive care: Rotavirus
vaccine; hygiene and
diapering precautions in
day care facilities.
Salmonella spp. 1 to 3 days
Diarrhea, fever,
abdominal cramps,
rebound tenderness,
vomiting. S.
typhi and S.
paratyphi produce
typhoid with
insidious onset
characterized by
fever, headache,
4 to 7 days Contaminated
eggs,
poultry,
unpasteurize
d milk or
juice, cheese,
contaminate
d raw fruits
and
Routine stool
cultures;
positive
leukocytes
and gross
blood. CBC:
WBC can be
slightly ↑
with left
Supportive
care: Only consider
antibiotics (other than
for S. typhi or S.
paratyphi) for infants <3
months old, those with
chronic GI disease,
malignant neoplasm,
hemoglobinopathies,
HIV, otherconstipation, malaise,
chills, and myalgia;
diarrhea is
uncommon, and
vomiting is not
usually severe
vegetables
(alfalfa
sprouts,
melons)
S. typhiepidemics
are often
related to
fecal
contaminatio
n of water
supplies or
streetvended
foods
shift, ↓, or
normal.
immunosuppressive
illnesses or therapies.
If indicated, consider
ampicillin or amoxicillin,
azithromycin, or TMPSMX; if resistance shown
to any of those, use IM
ceftriaxone, cefotaxime;
or azithromycin or
quinolones.
A vaccine exists for S.
typhi in certain cases.
Etiology Incubation
Period
Signs and
Symptoms
Duration
of
Illness
Route of
Transmission Laboratory Testing Treatment*
Cryptosporidium
parvum
3 to 14
days
Diarrhea
(usually
watery),
stomach
cramps,
upset
stomach,
bloating,
slight fever,
anorexia,
weight loss,
flatulence,
nausea,
vomiting,
fatigue
Selflimit
ing,
usua
lly
lasts
6 to
14
days
Fecal-oral route;
from uncooked
food or food
contaminated
by an ill food
handler after
cooking;
drinking water
(collects on
water filters and
membranes that
cannot be
disinfected);
reservoirs
include cattle,
sheep, goats,
birds, reptiles,
young animals
Request specific testing of
the stool
for Cryptosporidium usi
ng antigen-detection
tests. May need to
examine water or food.
Supportive care, selflimited. If severe, or
individual
immunocompromised
consider
paromomycin for 7
days. For children 1 to
11 years old, consider
nitazoxanide for 3
days.
Pyloric Stenosis
Pyloric stenosis is characterized by hypertrophied pyloric muscle, causing a narrowing of the pyloric sphincter. Pyloric stenosis occurs in 3
per 1000 live births, with a fourfold increase in males compared with females (Hunter and Liacourus, 2011b). It tends to be familial and is
seen more commonly in Caucasian first-born males.
Clinical Findings
History.
• Regurgitation and non-projectile vomiting during the first few weeks of life
• Projectile vomiting beginning at 2 to 3 weeks old
• Insatiable appetite with weight loss, dehydration, and constipation
• An association of pyloric stenosis with the administration of erythromycin in the first 2 weeks of life has been demonstratedPhysical Examination.
• Weight loss
• Nonbilious vomitus that can contain blood
• A distinct ―olive‖ mass that is often palpated in the epigastrium to the right of midline
• Reverse peristalsis visualized across the abdomen
Diagnostic Studies.
Ultrasound, with measurement of the pyloric muscle thickness, is used in most centers. An upper gastrointestinal series demonstrates a
―string sign,‖ indicating a fine, elongated pyloric canal may be required if ultrasound is unavailable or inconclusive.
Management and Prognosis
Surgical intervention (pyloromyotomy) is indicated after correction of fluid and electrolyte imbalance. Vomiting can continue for a few days
after surgery, although it is not as significant as it was preoperatively; feedings should be introduced gradually. The prognosis is excellent.
Intussusception
Intussusception involves a section of intestine being pulled antegrade into adjacent intestine with the proximal bowel trapped in
the distal segment. The invagination of bowel begins proximal to the ileocecal valve and is usually ileocolic, but it can be
ileoileal or colocolic. Intussusception is 859thought to be the most frequent reason for intestinal obstruction in children.
Intussusception most commonly occurs between 5 and 10 months of age and is also the most common cause of intestinal
obstruction in children 3 months to 6 years old; 80% of the cases occur before 2 years of age. In younger infants, intussusception
is generally idiopathic and responds to nonoperative approaches. In some children, there is a known medical predisposing
factor, such as polyps, Meckel diverticulum, Henoch-Schönlein purpura, constipation, lymphomas, lipomas, parasites, rotavirus,
adenovirus, and foreign bodies. Intussusception may also be a complication of CF. Children older than 3 years are more likely to
have a lead point caused by polyps, lymphoma, Meckel diverticulum, or Henoch-Schönlein purpura; therefore, a cause must be
investigated. The currently approved rotavirus vaccines have not been associated with an increased risk of intussusception
(Kennedy and Liacouras, 2011).
Clinical Findings
History
• The classic triad for intussusception, intermittent colicky (crampy) abdominal pain, vomiting, and bloody mucous stools, are present in
fewer than 15% of cases (Kennedy and Liacouras, 2011):
• Paroxysmal, episodic abdominal pain with vomiting every 5 to 30 minutes. Vomiting is nonbilious initially. Some children do not have
any pain.
• Screaming with drawing up of the legs with periods of calm, sleeping, or lethargy between episodes.
• Stool, possibly diarrhea in nature, with blood (―currant jelly‖).
• A history of a URI is common.
• Lethargy is a common presenting symptom.
• Fever may or may not be present; can be a late sign of transmural gangrene and infarction.
• Severe prostration is possible.
Physical Examination
• Observe the baby's appearance and behavior over a period of time; often the child appears glassy-eyed and groggy between episodes, almost
as if sedated.
• A sausage-like mass may be felt in the RUQ of the abdomen with emptiness in the RLQ (Dance sign); observe the infant when quiet
between spasms.
• The abdomen is often distended and tender to palpation.
• Grossly bloody or guaiac-positive stools.
Diagnostic Studies
• An abdominal flat-plate radiograph can appear normal, especially early in the course and reveal intussusceptions in only about 60% of cases
(Fig. 33-5). A plain radiograph may show sparse or no intestinal gas or stool in the ascending colon with air-fluid levels and distension in
the small bowel only.
• Abdominal ultrasound is very accurate in detecting intussusception and is the test of choice (Ross and LeLeiko, 2010). It shows ―target
sign‖ and the ―pseudo kidney‖ sign and can also be used to evaluate resolution following air contrast enema.• An air contrast enema is both diagnostic and a treatment modality.
Differential Diagnosis
The differential diagnosis includes incarcerated hernia, testicular torsion, acute gastroenteritis, appendicitis, colic, and intestinal obstruction.
860
Management
• Emergency management and consultation with a pediatric radiologist and a pediatric surgeon is recommended.
• Rehydration and stabilization of fluid status; gastric decompression.
• Radiologic reduction using a therapeutic air contrast enema under fluoroscopy is the gold standard.
• Surgery is necessary if perforation, peritonitis, or hypovolemic shock is suspected or radiologic reduction fails.
• IV antibiotics are often administered to cover potential intestinal perforation.
• A period of observation following radiologic reduction is recommended (12 to 18 hours); clear discharge instructions to return with any
recurrence of symptoms are required, and close phone follow-up for up to 72 hours is prudent.
Complications
Swelling, hemorrhage, incarceration, and necrosis of the bowel requiring bowel resection may occur. Perforation, sepsis, shock, and reintussusception (reported to typically be less than 10%, usually within 72 hours of radiologic reduction but can occur up to 36 months later)
can all occur. Recurrence is associated with the lead points described earlier.
Celiac disease is an immune-mediated systemic disorder triggered by dietary exposure to wheat gluten and related proteins in
barley and rye. It is characterized by the presence of a variable combination of gluten-dependent clinical manifestations, celiac
disease–specific antibodies, HLA-DQ2.5 or HLA-DQ8 haplotypes, and enteropathy. This disease frequently co-occurs with other
autoimmune diseases: diabetes mellitus type 1, autoimmune thyroiditis, autoimmune liver disease, IgA nephropathy, and
juvenile chronic arthritis (Mubarak et al, 2012). A number of conditions or variables may contribute to the development of celiac
disease. It is suggested that demographic changes, such as immigration from developing to developed countries, increase
exposure to gluten and an increased incidence of celiac disease follows (Scanlon and Murray, 2011). Celiac disease is greater
among infants born by cesarean section; the development of enteric homeostasis in the newborn period may be altered,
increasing susceptibility (Decker et al, 2010). Parent reported gastroenteritis occurring at the time gluten was introduced into the
child's diet does not appear to be associated with celiac disease (Welander et al, 2010). Celiac disease has a worldwide
distribution with overall prevalence of 1% (Mustalahti et al, 2010). The most typical presentation occurs between 6 months and 2
years old.
Clinical Findings
General History for Malabsorption Syndromes
Careful medical and family medical histories are very important in the evaluation of a malabsorption syndrome and are often
the key to the diagnosis. In addition, a complete dietary history is needed to distinguish between undernutrition and
malabsorption. Important historical findings include:
• Past surgical and trauma history
• Growth failure (a common symptom of nutritional deficiency and malabsorption)
• Delayed puberty can coexist with malabsorption.
• A voracious appetite or particular food avoidance is present in small children with malabsorption syndromes
• Chronic diarrhea with frequent, large, foul-smelling, pale stools
• Excessive flatus with abdominal distention
• Pallor, fatigue, hair and dermatologic abnormalities, digital clubbing, dizziness, cheilosis, glossitis, peripheral neuropathy
(symptoms of vitamin deficiency seen with malabsorption)
Disease-Specific History
In addition to the list in the earlier section, the following may stand out.
Celiac Disease.
• Chronic or intermittent diarrhea, persistent or unexplained GI symptoms (e.g., nausea and vomiting), sudden or unexpected weight loss, and
prolonged fatigueDiagnostic Studies
• Stool assessment for occult blood, WBCs, and culture; liquid stool for pH and reducing substances; 72-hour fecal fat collection or Sudan
stain for stool fat
• Spot stool testing for alpha 1-antitrypsin level to establish the diagnosis of protein-losing enteropathy
• Sweat chloride test (in the presence of steatorrhea to evaluate for CF)
• Stool for O&P: Giardiasis is a common intestinal infection causing malabsorption. See later discussion for symptoms suggestive of
infestation.
• CBC with differential, mean corpuscular hemoglobin concentration (MCHC), iron, folic acid, and ferritin
• Serum calcium, phosphorus, magnesium, alkaline phosphatase, serum protein, liver function tests, vitamin D and its metabolites, vitamins
A, B12, E, and K
• Human immunodeficiency virus (HIV) testing (for symptoms of FTT and chronic diarrhea)
• Small bowel biopsy helps identify diseases of the small bowel mucosa and obtain material for culture and sensitivity
• Plain abdominal radiographs and barium contrast studies as indicated
• Abdominal ultrasound can detect masses and stones in the hepatobiliary system
• Retrograde studies of the pancreas and biliary tree if indicated
• Bone age
Specific Tests for Celiac Disease
• Serologic testing should be done if there is clinical suspicion of celiac disease, the child has an associated disorder, or there is a first-degree
relative with celiac disease. Gluten should be eaten in more than one meal every day for 6 weeks prior to testing. Recommended serologic
tests include IgA tissue transglutaminase antibody (tTGA) and IgA endomysial antibody (EMA) because of their high sensitivity and
specificity (Guandalini et al, 2014; Tran, 2014). EMA is more expensive and less accurate in children younger than 2 years old (Gelfand,
2013).
• Home blood testing is not recommended (NICE, 2010).
• If serologic testing is positive, refer for endoscopy with biopsy for a definitive diagnosis, although colonoscopy may not be necessary if the
tTGA level is greater than 100 units/mL (Mubarak et al, 2011).
• Careful follow-up of growth parameters, tTGA testing after 6 months of gluten-free diet (GFD), and then yearly (Hill et al, 2005).
• Bone density testing (bone problems may be first symptom of celiac disease).
Management
Celiac Disease
• A strict GFD for life is currently the only effective treatment for celiac disease. The standard for being gluten-free is a limit of
20 ppm of gluten (Hill et al, 2005). Adding pure oats to a GFD can improve palatability and increase fiber and vitamin B intake
without causing a systemic or autoantibody response (Mubarak et al, 2012; Scanlon and Murray, 2011).
• Alternative treatments are being explored, including enzyme therapy, developing genetically engineered grains, inhibiting
tTGA in the intestine, and correcting intestinal barrier defects (particularly increased permeability).
• A lactose-free diet for young children may be helpful. This is generally not the case in adolescents and adults unless they are
lactose intolerant (Hill et al, 2005).
Complications and Prognosis
Celiac Disease
Growth failure is the primary complication of celiac disease. With delayed diagnosis or inadequate treatment, there is risk for
fractures and osteoporosis (due to reduced bone mineral density), lymphoma, autoimmune diseases (e.g., type 1 diabetes,
thyroid disorders), primary biliary cirrhosis, and primary sclerosing cholangitis. Sensory peripheral neuropathy may be related
to gluten sensitivity (Hadjivassiliou et al, 2010). Celiac crisis consisting of abdominal distention, explosive watery diarrhea,
dehydration with hypoproteinemia, electrolyte imbalance, hypotensive shock, and lethargy, although rare, can be the first
indication of celiac disease. Prognosis is improved with lifelong GFD.
Juvenile Idiopathic Arthritis
JIA, formerly known as juvenile rheumatoid arthritis (JRA), now encompasses several disorders that have a common feature of
arthritis (e.g., enthesitis-related arthritis and psoriatic arthritis) and had not been identified under the nomenclature of JRA (Wuet al, 2011). The diagnosis of JIA requires a persistent arthritis for more than 6 weeks in a pediatric patient younger than 16 years
old. Table 25-1 shows the most current classification system.
TABLE 25-1
Juvenile Idiopathic Arthritis Subtypes and Clinical Joint Characteristics
Juvenile Idiopathic Arthritis
Subtype Clinical Joint Characteristics
Oligoarticular Four or less joints with persistent disease never having more than four-joint involvement and extended disease progressing
to more than four joints within the first 6 months
Polyarticular (RF negative) Five or more joints with symmetrical involvement
Polyarticular (RF positive) Symmetric involvement of both small and large joints with erosive joint disease
Systemic Either polyarticular or oligoarticular disease
Enthesitis-related arthritis Weight-bearing joints involved especially hip and intertarsal joints and a history of back pain, which is inflammatory in nature
or sacroiliac joint involvement
Psoriatic arthritis Asymmetric or symmetric small or large joints
Undifferentiated
RF, Rheumatoid factor.
The underlying cause of most forms of JIA is unclear; however, it is a heterogenous disorder. It is likely environmentally
induced in genetically predisposed individual. Human leukocytic antigen (HLA) class I and II alleles have been associated with
JIA (Gowdie and Tse, 2012). This linkage points to the involvement of T cells and antigen presentation in the pathophysiology of
the disease. An environmental trigger, such as infection or trauma, is also important in the pathogenetic process in JIA. The
trigger results in an uncontrolled adaptive and innate response toward the self-antigen, the autoimmune reaction. The presence
of autoantigens from cartilage and joint tissue leads to activation of the T cells and results in release of proinflammatory
cytokines (Gowdie and Tse, 2012). In contrast, systemic juvenile idiopathic arthritis (SJIA), which does not have HLA gene
association, may be the result of an autoinflammatory response from the innate immune system. SJIA is postulated to be the
result of uncontrolled activity of the innate immune system, because this type of JIA disease is not associated
with 552autoantibodies but rather uncontrolled activity of the phagocytes, including neutrophils, monocytes, and macrophages.
The difference in the pathogenic processes may explain the differences in the clinical presentation of the disease.
In oligoarticular and rheumatoid factor (RF)-positive polyarticular JIA, there is autoimmunity with involvement of the
adaptive immune system. The presence of positive ANAs and RF is associated with HLA genes. The humoral response is
responsible for the release of autoantibodies (especially ANAs), an increase in serum immunoglobulins, and the formation ofcirculating immune complexes and complement activation. The cell-mediated reaction is associated with a T-lymphocyte
response that plays a key role in cytokine production, resulting in the release of tumor necrosis factor alpha (TNF-α), IL-1, and
IL-6. B lymphocytes are activated by T-helper cells and produce autoantibodies that link to self-antigens. The B lymphocytes
infiltrate the synovium with the end result of nonsuppurative chronic inflammation of the synovium that can lead to articular
cartilage and joint structure erosion.
The chronic arthritides of childhood present unique challenges to the child, family, and the pediatric provider.
Approximately 1 in 1000 children are affected with oligoarticular JIA, the most common arthritic subtype. Certain
histocompatibility complex antigens are more prevalent in the JIA population. Cytokine production, proliferation of
macrophage-like synoviocytes, infiltration with neutrophils and T lymphocytes, and autoimmunity are thought to be the major
pathologic processes causing chronic joint inflammation.
The rate of JIA is significantly higher in girls than in boys, typically in oligoarticular and pauciarticular JIA. The female to
male ratio in systemic onset is equal. The approximate percentage of occurrence and age breakdown for each of the subtypes
follows: systemic (10%) occurs at any age; polyarticular (40%) has a late (6 to 12 years old) or early childhood (1 to 4 years old)
onset; and oligoarticular (50%) has a late or early onset. Adolescents tend to have more RF-positive disease (Wu et al, 2011).
Clinical Findings
History
The major complaints in all forms of JIA are from the arthritis characterized by:
• Pain—generally a mild to moderate aching
• Joint stiffness—worse in the morning and after rest; arthralgia may occur during the day
• Joint effusion and warmth
Systemic symptoms are found more commonly in systemic and polyarticular subtypes and include anemia, anorexia, fever, fatigue,
lymphadenopathy, salmon-colored rash (SJIA), and weight loss. Growth abnormalities can result in localized growth disturbances, including
premature fusion of the epiphyses, bony overgrowth (rheumatoid nodules), and limb-length discrepancies.
Physical Examination
Associated features are:
• Non-migratory monoarticular or polyarticular involvement of large or proximal interphalangeal joints for more than 3 months
• Systemic manifestations—fever, salmon-colored rashes, leukocytosis, serositis, lymphadenopathy, and rheumatoid nodules
Less commonly seen are ocular disease (e.g., iridocyclitis, iritis, or uveitis), pleuritis, pericarditis, anemia of chronic disease, fatigue, and
growth failure, or leg-length discrepancy if the arthritis is unilateral.
Key physical findings are:
• Swelling of the joint with effusion or thickening of synovial membrane, or both, noted on palpation of the joint line
• Heat over inflamed joint and tenderness along joint line
• Loss of joint range of motion and function; child typically holds the affected joints in slight flexion and may walk with limp
• Uveitis may be present with ciliary injection and decreased vision. However, it is usually asymptomatic.
There are five major types of JIA (Gowdie and Tse, 2012):
1. Oligoarticular pattern: This type of JIA involves four or less joints, typically the weight-bearing joints within the first 6 months of
diagnosis. The diagnosis is classified as persistent or extended disease, depending on the number of joints involved. About 50% progress
to extended disease where there is involvement of four or more joints after the first 6 months of disease. This involvement primarily is in
larger or medium joints, such as the knee, ankle, wrists, and elbow; however, systemic symptoms are rare. The synovitis may be mild and
painless with asymmetric joint involvement and unremarkable laboratory values. Uveitis occurs in 30% especially if the child has a
positive ANA (Gowdie and Tse, 2012).
2. Polyarticular pattern: This involves five or more joints and is divided into RF-negative and RF-positive disease. Involved joints can be
large or small with an acute or insidious onset. RF-negative ANA positive polyarticular JIA is difficult to distinguish from extended
oligoarticular pattern disease. Using the number of joints involved and the timing of onset of the arthritis can be helpful. In contrast, RFpositive disease can have chronic pain and symmetric joint swelling, low-grade fever, fatigue, nodules, and anemia of chronic disease. An
acute form of uveitis occurs in this subtype. Polyarticular JIA typically involves small joints of the hands, feet, ankles, wrists, knees, and
can also involve the cervical spine. Adolescents with this type differ from those with early onset in that they exhibit a positive RF.
Adolescents who develop late-onset polyarticular JIA have a course similar to the adult entity. Both forms of the disease are more
common in females.
3. SJIA: This is characterized by arthritis in one or more joints for 6 weeks' duration in a child younger than 16 years old with a fever of at
least 2 weeks' duration with 553at least 3 days of daily fever. In addition, there is also a fleeting erythematous rash, lymphadenopathy,
hepatomegaly, splenomegaly, and serositis (Ringold et al, 2013). Myocarditis with pericardial effusion occurs in approximately 10%. RF
is rarely positive and the ANA is only positive in 5% to 10%; however, there may be anemia, thrombocytosis, increased acute phase
reactants, and elevated transaminase levels. About 10% of children with SJIA develop a life-threatening macrophage activation syndrome(MAS) with fever, organomegaly, cytopenia, hyperferritinemia (acute phase reactant), hypertriglyceridemia, coagulopathy, and
hypofibrinogenemia.
4. Enthesitis-related JIA: This typically entails arthritis of the lower limbs especially the hip and intertarsal joints with the sacroiliac joints
involved later in the disease. Enthesitis involves inflammation at the insertion of tendons, ligaments, or joint capsules and is characterized
by swelling, tenderness, and warmth. Enthesitis may present with joint or foot pain. There is a risk of anklyosing spondylitis 10 to 15
years later. It tends to occur in late childhood and adolescence and acute symptomatic uveitis occurs in about 7%.
5. Psoriatic arthritis: This is more common between the ages of 2 and 4 and again between 9 to 11 years old. There is usually a family history
of psoriasis, or the child has psoriasis; however, the arthritis can precede the psoriasis by years. There can be dactylitis or a sausage-like
swelling of the digits; involvement in the small digits is not uncommon.
Diagnostic Studies
JIA is a diagnosis of exclusion. The diagnosis is based on physical findings and history of arthritis lasting for 6 weeks or longer. There is no
diagnostic laboratory test for JIA. Most children with oligoarticular arthritis have negative laboratory markers. Those with polyarticular and
systemic-onset typically have elevated acute-phase reactants and anemia of chronic disease. A positive result for RF by latex fixation may be
present, but a positive RF occurs in less than 10% of children with JIA and rarely in those with SJIA. ANA may be present in up to 50% of
children with oligoarticular disease. A positive ANA helps identify children at higher risk for uveitis. The anti-CCP antibody test can be
added to the initial workup of JIA, because citrullinated residues are part of the essential antigenic components that are recognized by
autoantibodies in rheumatoid arthritis (Mehta, 2012). The anti-CCP antibodies are associated with more aggressive disease and may be
present before the onset of symptoms. The anti-CCP antibody is highly specific, but its precise role has not been established because it is
found primarily in children with polyarticular JIA (Mehta, 2012). Useful laboratory tests include a complete blood count (CBC) (to exclude
leukemia); ESR, CRP, Lyme titers, and liver function tests. The results may reveal lymphopenia, anemia, elevated transaminases, and
hypoalbuminemia; however, laboratory studies may be normal in these children. Imaging studies (MRI) can help in managing joint
pathologic conditions. Analysis of synovial fluid is not helpful in the diagnosis of JIA.
Differential Diagnosis
The various causes of monoarticular arthritis are part of the differential diagnosis. However, Lyme disease must be excluded and other
differentials, including tumors, leukemia, cancer, bacterial infections, toxic synovitis, rheumatic fever, SLE, spondyloarthropathies,
inflammatory bowel disease, septic arthritis, and chondromalacia patellae, need to be carefully considered.
Management
A specialist in pediatric rheumatology should follow children with severe involvement. Ophthalmology referral and evaluation is critical in a
child with a positive ANA. Uveitis needs immediate ophthalmologic management. It is most common in oligoarticular JIA and is highly
associated with a positive ANA. Other pediatric subspecialists, such as orthopedists, pain management specialists, and cardiologists, may be
consulted as needed. Therapy depends on the degree of local or systemic involvement.
The main treatment goals are to suppress inflammation, preserve and maximize joint function, prevent joint deformities, and prevent
blindness. Drug therapy is used to control the inflammation responsible for tissue injury with the goal of preventing permanent tissue
changes, which is not always possible. Aggressive early treatment to induce a remission is a key consideration in JIA management in order to
prevent deformity and improve outcomes and is now the goal of the practice guidelines for both polyarticular JIA and SJIA (Ringold et al,
2013, 2014). Aspirin therapy has largely been replaced with the use of nonsteroidal anti-inflammatory drugs (NSAIDs). Pharmacologic
agents commonly used in the management of JIA include the following (Gowdie and Tse, 2012):
• NSAIDs: Children with oligoarthritis generally respond well to NSAIDs (Taketomo et al, 2014).
• Ibuprofen: 30 to 40 mg/kg/day three to four divided doses (maximum single dose is 800 mg; maximum daily dose 2400 mg/day)
• Tolmetin: 20 to 30 mg/kg/day divided in three to four doses (maximum dose is 1800 mg/day)
• Naproxen: 10 mg/kg/day in two divided doses (maximum dose is 1000 mg/day)
• Indomethacin: Older than 2 years old, 1 to 2 mg/kg/day divided in two to four doses (maximum dose is 4 mg/kg/day); adults, 25 to
50 mg/dose two or three times/day (maximum dose is 200 mg/day)
• Celecoxib: Older than 2 years old and adolescents (≥10 kg to ≤25 kg), 50 mg twice daily; >25 kg, 100 mg twice daily
• Oral, parenteral, intraarticular corticosteroids:
• Systemic arthritis: Can be used for 2 weeks as initial therapy for SJIA with involvement of more than four joints and a physician global
assessment (using the Provider global assessment tool of disease activity) of 554less than 5 or a Provider global score of more than 5
without care about active joint involvement. Corticosteroids can be used as bridging therapy until other medications take effect
(Ringold et al, 2013)
• All the other types of arthritis: Prednisone in the lowest possible dose with optional intraarticular steroid injection (Ringold et al, 2014)
• Disease-modifying antirheumatic drugs (DMARDs): Recent published guidelines vary related to the initiation of these agents depending on
type of arthritis, joint involvement, and MD global assessment of functioning
• Nonbiologic DMARD treatment: methotrexate, sulfasalazine, leflunomide (managed by pediatric rheumatologist)
• Biologic DMARD treatment (managed by pediatric rheumatologist)
• Short-acting agents: Anti-IL-1 anakinra is the first-line agent for SJIA with significant joint involvement and poor global
functioning (Sterba and Sterba, 2013).
• Long-acting agents: Rilonacept, canakinumab, and tocilizumab have long-acting activity (Sterba and Sterba, 2013). Rilonacept is a
recombinant fusion protein with high affinity for IL-1β, IL-1α, and IL-1 receptors and a half-life of 8.6 days. Canakinumab is ahumanized monoclonal antibody effective against IL-β with a half-life of 28 days. Tocilizumab is effective against IL-6 (Sterba
and Sterba, 2013).
• TNF-α agents: For example, etanercept (Enbrel, infliximab (Remicade), and adalimumab (Humira) soak up tumor necrosis factor,
an immune-system protein, and block the inflammatory cascade. Methotrexate or anakinra is used in severe forms of JIA.
• Intraarticular corticosteroid injections are used if there is severe joint involvement.
• Pharmacologic therapy for uveitis is given as indicated by an ophthalmologist. Females with ANA-positive oligoarticular JIA are at
high risk for uveitis and require slit-lamp examination every 3 to 4 months. The uveitis often does not correspond to the severity of
the arthritis (i.e., uveitis may be present despite quiescent arthritis).
• Physical therapy—range of motion muscle-strengthening exercises and heat treatments—is used for joint involvement; occupational
therapy is beneficial. Rest and splinting are used if indicated.
• Ophthalmologic follow-up every 3 months for 4 years (even if the arthritis has resolved) for all ANA-positive JIA children. They
have a greater risk of uveitis that may not be clinically apparent but can lead to blindness if not detected and treated.
Complications and Prognosis
Systemic involvement can include iridocyclitis, uveitis, pleuritis, pericarditis, anemia, fatigue, and hepatitis. Residual joint damage caused by
granulation of tissue in the joint space can occur. Children most likely to develop permanent crippling disability include those with hip
involvement, unremitting synovitis, or positive-RF test.
The course of the disease is variable, and there is no curative treatment. Again, early aggressive treatment is critical; therefore, referral to a
specialist is important. After an initial episode, the child may never have another episode, or the disease may go into remission and recur
months or years later. The disease process of JIA wanes with age and completely subsides in 85% of children; however, systemic onset, a
positive RF, poor response to therapy, and the radiologic evidence of erosion are associated with a poor prognosis. Onset of disease in the
teenage years is related to progression to adult rheumatoid disease.
Patient and Parent Education and Prevention
The following education and preventive measures are taken:
• For children on aspirin therapy (not typically given), educate parents about the risk of Reye syndrome and its signs and symptoms.
• Recommend yearly influenza vaccine.
• Offer chronic disease counseling and encourage normal play and recreation.
• Educate about side effects of medications, in addition to splinting, orthotics, and bracing requirements.
• Instruct about need to follow up with an ophthalmologist. Frequency of follow-up for uveitis screening is based on subtype of JIA and is
determined by protocol guidelines and ophthalmology.
• Ensure parent and child understand that physical therapy is a mainstay of treatment for chronic childhood arthritis and should be part of the
child's daily routine. A daily plan should include passive, active, and resistive exercises.
• Water therapy and the use of heat or cold reduce pain and stiffness. Swimming is an excellent activity except for children with severe
anemia and severe cardiac disease.
• Tricycle or bike riding and low-impact dance are other beneficial activities.
• Refer to the American Arthritis Foundation and the Juvenile Arthritis Association, which have excellent resources for family members and
children.
• Instruct on the need to involve school personnel in the identification of required school-related services through an individualized education
plan (IEP) or a 504.
• Discuss the challenge of pain management and its assessment in children with chronic arthritis and encourage parents to advocate for
effective pain control on behalf of their child.
Puncture Wounds (Osteomyelitis)
Description and Epidemiology
Puncture wounds result from penetration of varying levels of skin and underlying tissue. These wounds are typically classified as superficial
or deep. Glass, wood splinters, toothpicks, needles, nails, metal, staples, thumbtacks, and bites are common sources of injury. Although the
majority of puncture wounds heal without problems, a sizable minority of these injuries are complicated by infections that can lead to
cellulitis, fasciitis, septic arthritis, or soft-tissue abscesses.
Staphylococcus aureus and beta-hemolytic streptococci are normal flora of the skin and are common causes of secondary infections in
puncture wounds. Pseudomonas aeruginosa colonizes on the rubber soles of tennis shoes and is a common pathogen for plantar puncture
wounds when the puncture occurs through the sole of a tennis shoe and into the foot. Osteomyelitis can occur if the puncture wound
penetrates a bone or joint and is most commonly caused by P. aeruginosa in nondiabetic patients and is most commonly caused by S.
aureus in diabetic patients (Baddour, 2013). Cat and dog bites can cause wound infection from Pasteurella multocida.
When considering risk for infection, the location and depth of the wound and the presence of a foreign object are important components.
For example, deep penetrating injuries to the forefoot with a dirty object, especially if they involve the plantar fascia, have a higher risk ofinfection than wounds to the arch or heel area. The forefoot has less overlying soft tissue than other plantar surfaces and is the major weightbearing area of the foot; therefore, cartilage and bone can be involved. The metatarsophalangeal joint region is also at high risk for infection
for the same reasons. Puncture wounds through the soles of tennis shoes can transfer bacteria into the tissue while simultaneously impairing
wound drainage, placing the child at higher risk for a secondary infection.
Assessment
The assessment of a child with a minor wound begins by excluding more serious and sometimes occult injuries.
History.
Important information to elicit after a report or suspicion of a puncture wound includes the following:
• Date and time of injury and history of wound care provided at time of injury and thereafter.
• Identification of the penetrating object and the type and estimated depth of penetration. If it is not known what object penetrated the skin,
the likelihood of an imbedded foreign body is high.
• Location and condition of the penetrating object. Was the object clean or rusty, jagged or smooth?
• Whether all or part of the foreign object was removed.
• Type and condition of footwear that was being worn (pertinent to injuries to the foot) or if the child was barefoot.
• Immunization status for tetanus coverage (see Chapter 24).
• Presence of any medical condition that increases the risk for infectious complications.
Physical Examination.
A good light source is necessary to assess and treat a puncture wound. Note circulation, movement, and sensation of the area next to the
injury. Determine the amount of involvement of underlying tissue or bone structures. For plantar puncture wounds, have the patient lie prone
with the feet positioned at the head of the examining table and the knees slightly flexed (Buttaravoli and Leffler, 2012). Assess the wound for
length and depth, presence of debris or penetrating object, and signs of infection.
Examination findings consistent with cellulitis include:
• Localized pain or tenderness, swelling, and erythema at the puncture site (may be more obvious at dorsum of the foot for plantar puncture
wounds)
1127
• Possible fever
• Pain with flexion or extension of the extremity involved
• Decreased ability to bear weight
• For plantar puncture wounds, pain along the plantar aspect of the foot during extension or flexion of the toes may indicate deep tissue injury,
thus a higher risk of infection
Examination findings consistent with osteomyelitis-osteochondritis include:
• Extension of pain and swelling around the puncture wound and to the adjacent bony structures
• Exquisite point tenderness over the bone
• Fever
• Increasing erythema
• Decreased use of the affected extremity
Examination findings consistent with pyarthrosis (septic arthritis) include:
• Pain, swelling, warmth, and erythema over the affected joint
• Decreased range of motion and weight bearing of the affected joint
• Fever
Diagnostic Studies.
Plain film radiograph should be ordered if any of the following occur:
• A suspicion of a retained foreign object.
• There is a tremendous amount of pain at the site of the wound, localized tenderness is noted over the wound, there is discoloration
underneath the skin surface, or there is a palpable mass noted at or near the wound entry site (Baddour, 2013).
• There was penetration of a joint space, bone or growth cartilage, or the plantar fascia of the foot.
• The puncture site has signs of infection and is from a nail injury.
• Most metal and glass foreign bodies can be seen on a plain radiograph. However, if the foreign object is not radiopaque or if the x-ray
is negative despite suspicion of foreign object in the wound, computed tomography (CT), ultrasound, and magnetic resonance
imaging (MRI) are useful diagnostic tools (Buttaravoli and Leffler, 2012).• Bone scans are sensitive but not specific for osteomyelitis. Radiographs are specific, but findings for osteomyelitis are noted late.
Clinical examination and laboratory studies and imaging should be considered early in the diagnosis of osteomyelitis (Erickson and
Caprio, 2014).
• A complete blood count (CBC) and blood culture may be needed. An elevation in the white blood cell count might indicate infection.
• An erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are nonspecific inflammatory markers and are helpful in the
diagnosis and management of bony inflammation and infection.
• A wound culture is indicated prior to starting antibiotics if the wound appears infected.
Differential Diagnosis
The history and physical examination provide the diagnosis.
Management
The circumstance surrounding the penetrating injury and the presenting symptoms are the best indicators of whether the injury is superficial
and will heal uneventfully or if it will result in infectious complications. Buttaravoli and Leffler (2012) suggest the following practical and
straightforward approach to the management of puncture wounds:
• For the majority of superficial or simple puncture wounds, débridement with an antiseptic solution after scrubbing the wound surface is
sufficient. Consider the use of wound irrigation. Ensure that there are no foreign bodies present.
• A No. 10 scalpel may be used to gently shave off the cornified epithelium surrounding the puncture wound to aid in the removal of debris
that collected around the point of entry.
• For wounds where debris is noted, gently slide the plastic sheath of an over-the-needle catheter down the wound track and move the catheter
sheath in and out while irrigating with copious amounts of normal saline until debris no longer flows from the wound. A local anesthetic
agent may be necessary for débridement and irrigation procedures.
• Obtain imaging studies as indicated. If imaging studies demonstrate that the foreign object has invaded bone, growth cartilage, or a joint
space, refer the child immediately to an orthopedic surgeon. Always suspect a retained foreign object if the puncture wound is infected, the
infection is not responding to antibiotic therapy, or if pain or aching of the injured site is still present weeks after the injury. In order to
prevent a catastrophic outcome, wounds that are deep or highly contaminated should be referred to an orthopedic surgeon so that
débridement can take place in an operating room (Buttaravoli and Leffler, 2012).
• Following careful wound cleansing, the wound can be covered with a simple bandage. Deeper wounds that require more extensive
exploration should have a small sterile wick of iodoform gauze placed in the wound track in order to keep the edges open, thus aiding in
granulation tissue growth and wound healing. Remove the gauze 2 to 3 days after placement (Selbst and Attia, 2010).
• Children with simple, uncomplicated puncture wounds do not need antibiotics; however, if there are signs of infection, the puncture is the
result of a cat bite, or if the wound is deep or contained debris, antibiotics should be part of the treatment plan. Appropriate antibiotics for
puncture wounds include amoxicillin clavulanate or cephalexin. Clindamycin should be used when children are allergic to penicillins.
Plantar puncture wounds require ciprofloxacin. If methicillin-resistant Staphylococcus aureus (MRSA) is cultured from the wound or pus is
present at the puncture site, then trimethoprim-sulfamethoxazole (TMP-SMX) 1128or clindamycin is recommended until sensitivities are
known. All antibiotics should be prescribed for 7 to 14 days depending on severity of infection (Baddour, 2013). A recheck appointment
should be scheduled 48 hours from the start of antibiotics for the patient receiving outpatient therapy.
• Surgical débridement for removal of a foreign body and/or abscess drainage should be considered with an infected puncture wound
(Baddour, 2013).
• Treatment for severe infections secondary to puncture wounds, such as septic arthritis and osteomyelitis, includes surgical débridement and
parenteral antibiotics (Hosalkar et al, 2011).
• Tetanus prophylaxis is indicated if it has been more than 5 years since the last tetanus vaccine or if the date of the last tetanus vaccine is
unknown. Consider passive immunization with tetanus immune globulin (TIG) or initiation/continuation of a primary tetanus series (DTaP,
Tdap, or Td as appropriate) for children who have never been immunized or are behind in their vaccinations (see Chapter 24).
Patient and Parent Education
Home care management for a puncture wound includes:
• Cleanse the wound two times a day and when soiling of the wound occurs. Use warm water and soap, and then apply bacitracin or triple
antibiotic ointment to the wound.
• Cover the wound with a dressing, such as an adhesive bandage.
• Observe closely for signs and symptoms of infection and if infection is suspected, notify the provider immediately; rapid reevaluation is
necessary. Further evaluation is required if a puncture wound continues to cause localized or spreading pain or discomfort.
Transient
synovitis
3 to 8
years
+ Mild to moderate fever, mild
irritability; resolves within 1
Limited hip motion;
ESR <25 mm/h
Inflammatory reaction; unknown
etiology; often URI (50%) prior
Restold week
Hip Problems
Developmental Dysplasia of the Hip
Developmental dysplasia of the hip (DDH) represents a spectrum of anatomic abnormalities in which the femoral head and the acetabulum
are in improper alignment and/or grow abnormally. This includes dysplastic, subluxated, dislocatable, and dislocated hips. Dysplasia is
characterized by a shallow more vertical acetabular socket with an immature hip/acetabulum. In subluxation, the hip is unstable, and the head
of the femur can slide in and out of the acetabulum. DDH occurs congenitally or develops in infancy or childhood. Dysplasia may be
diagnosed many years after the newborn period.
Physiologic, mechanical, and genetic factors are implicated in DDH. Physiologic factors include the hormonal effect of maternal estrogen
and relaxin that are released near delivery and produce a temporary laxity of the hip joint. Mechanical factors include constant compression
in utero 1061with restriction of movement late in gestation if the fetal pelvis becomes locked in the maternal pelvis. This is seen with first
pregnancy, oligohydramnios, and breech presentation. The incidence of DDH is greater than normal in cultures that swaddle infants in
extended position or place them on cradleboards because of such neonatal positioning.
In the unstable hip, the femoral head and the acetabulum may not have a normal tight, concentric anatomic relationship, which can lead to
abnormal growth of the hip joint and result in permanent disability. In the newborn, the left hip is most often involved because this hip
typically is the one in a forced adduction position against the mother's sacrum.
The hip can dislocate non-congenitally or in utero in children with certain muscular or neurologic disorders that affect the use of the lower
extremities, such as cerebral palsy, arthrogryposis, or myelomeningocele. Dislocation results from the abnormal use of the extremity over
time.
The incidence of DDH is estimated to range from 1.5 to 20 per 1000 live births in the United States. It is found more commonly with
breech births and is four times more common in girls than boys. A positive family history (genetic risk factors) increases the risk for having a
child with this problem. Other risk factors seen in infants that are associated with DDH include oligohydramnios, torticollis, and lower limb
deformities, such as clubfoot, MA, and dislocated knee.
Clinical Findings
History.
Risk factors for DDH include female gender, family history, high birth weight, breech positioning, and in utero postural deformities (Herring,
2014a).
Physical Examination.
A hip examination should be performed on children as part of their well child supervision until they are 2 years old. Findings of DDH include
the following:
• Screening tests are serial physical examinations of the hip and lower extremities. The Barlow and Ortolani tests are used to screen for DDH
in neonates. Once an infant reaches the 2nd and 3rd months of life, the soft tissue surrounding the hips begins to tighten and the Barlow and
Ortolani tests are less reliable. The Klisic and Galeazzi tests are used to screen older infants. Routine ultrasonography is not recommended;
however, an ultrasound should be obtained if there is a high index of suspicion of dysplasia based on a positive clinical examination.
• Sixty percent to 80% of abnormal hips of newborns identified by physical examination resolve by 2 to 8 weeks.
• In the older infant, 6 to 18 months old:
• Limited abduction of the affected hip and shortening of the thigh is a reliable sign (see Fig. 38-3, B, and Fig. 38-5).
• Normal abduction with comfort is 70 to 80 degrees bilaterally. Limited abduction includes those cases with less than 60 degrees of
abduction or unequal abduction from one side to the other (see Fig. 38-5).
• Positive Galeazzi sign (see Fig. 38-3, A).
• Other findings include asymmetry of inguinal or gluteal folds (thigh-fold asymmetry is not related to the disorder [see Fig. 38-3, D]) and
unequal leg lengths, shorter on the affected side.
In the ambulatory child who was not diagnosed earlier or was not corrected, the following might also be noted:
• Short leg with toe walking on the affected side
• Positive Trendelenburg sign (see Fig. 38-3, C)
• Marked lordosis or toe walking
• Painless limping or waddling gait with child leaning to the affected side
If the hips are dislocated bilaterally, asymmetries are not observed. Limited abduction is the primary indicator in this situation (see Fig.
38-5). Also in subluxation of the hip (not frankly dislocated), limited abduction again is the primary indicator. A waddling gait may also be
noted.Diagnostic Studies.
Ultrasound is superior to radiographs for evaluating cartilaginous structures and is recommended for infants after 4 weeks of age. Use of
ultrasonography prior to 4 weeks old has a high incidence of producing false-positive results. Ultrasound is used to assess the relationship of
the femur to the acetabulum and provides dynamic information about acetabular development and stability of the hip. Radiologic evaluation
of the newborn to detect and evaluate DDH is recommended once the proximal epiphysis ossifies, usually by 4 to 6 months (Sankar et al,
2011). Radiography prior to this is unreliable, because so much of the hip joint is cartilaginous in the young infant. AP and lateral Lauenstein
(frog-leg) position radiographs of the pelvis are indicated.
Differential Diagnosis
The condition is relatively unique.
Management
The goal of management is to restore the articulation of the femur within the acetabulum. Many newborns with positive screening tests and
abnormal hips resolve without intervention; however, prompt referral to an orthopedist is important. The orthopedist needs to reexamine the
newborn to determine whether early treatment is necessary.
• The majority of neonatal hip instability cases resolve spontaneously by 6 to 8 weeks old. Close observation of these children is
recommended.
• The treatment of choice for subluxation and reducible dislocations identified in the early phase is a Pavlik harness. The harness is applied
with hips having greater than 90 degrees of flexion and with adduction of the hip limited to a neutral position. The success rate of Pavlik
harness treatment is reported to be between 80% and 97%. Radiographic or ultrasound documentation can be used during treatment to
verify the position of the hip. If the infant does not respond to treatment with the harness, surgical treatment may be needed.
1062
• The earlier that treatment is started with the Pavlik harness, the better the prognosis for a successful outcome. The harness is worn 24 hours
a day, except for bathing. The infant with a Pavlik harness should be seen weekly to ensure it fits properly, to identify complications
associated with the use of the harness (e.g., avascular necrosis and femoral nerve palsy), and to ensure the femur is properly seated in the
socket. Ultrasonography can be performed while the Pavlik harness is worn to assess hip reduction and acetabular development. The length
of time the harness is worn depends on the age of the child, when it was applied, and whether or not reduction is successful. Generally the
harness is worn full time for 3 to 6 weeks and then may be required only during waking hours for decreasing periods of time.
• For a child in a Pavlik harness or spica cast, cast care, skin care, and car safety when the child cannot easily be placed in a car seat are issues
to be addressed. Furthermore, the child needs special attention to maintain developmental stimulation while immobilized. An orthopedist
should be immediately consulted for any infant seen in a primary care setting who is in a Pavlik harness and exhibits excessive hip flexion
(beyond 100 degrees) or abduction (beyond 60 degrees).
• The 6- to 18-month-old infant with a dislocated hip is likely to require either closed manipulation or open reduction. Preoperative traction,
adductor tenotomy, and gentle reduction are especially helpful in preventing osteonecrosis of the femoral head. After the closed or open
reduction, a hip spica cast is applied in order to maintain the hip in more than 90 degrees of flexion and avoid excessive internal or external
rotation (Herring, 2014a). Triple diapering is not helpful, because the musculoskeletal forces far outweigh the force that can be exerted by
the diaper material.
• Annual or biennial follow-up including radiographs to the point of skeletal maturity is recommended to evaluate for the possibility of late
asymmetric epiphyseal closure (Herring, 2014a). Support the child and family through the treatment phases. Explain management goals
clearly.
Complications
The Pavlik harness and other positional devices may cause skin irritation, and a difference in leg length may remain. There may be delay in
walking if the child is put in a body cast. The long-term outcomes depend on the age at diagnosis, the severity of the joint deformity, and the
effectiveness of therapy. Untreated cases may result in a permanent dislocation of the femoral head so that it lies just under the iliac crest
posteriorly. Clinically, the child has limited mobility of this pseudo joint and related short leg. Forceful reduction can result in avascular
necrosis of the femoral head with permanent hip deformity. Redislocation or persistent dysplasia can occur. Adult degenerative arthritis is
associated with acetabular dysplasia.
Prevention
The condition cannot be prevented, but early identification resulting in early treatment significantly reduces the long-term consequences of
the problem. Screening of all neonates and infants should include full hip abduction; examination for unequal inguinal and gluteal folds and
unequal leg lengths; and Barlow, Ortolani, and Galeazzi maneuvers at every examination. The hip can dislocate at any point in early
development, even up to the point of first ambulation. In older children, limited abduction, gait, and standing position, including the
Trendelenburg position, add important information. Charting should always include notation about hip findings, because these can change at
subsequent visits. The Ortolani test should only be used in the first 2 to 3 months of age.
Legg-Calvé-Perthes Disease
LCPD is a childhood hip disorder that results in infarction of the bony epiphysis of the femoral head. It presents as avascular necrosis of the
femoral head. The basic underlying cause of LCPD is insufficient blood supply to the femoral head. There is an initial ischemic episode ofunknown etiology that interrupts vascular circulation to the capital femoral epiphysis. The articular cartilage hypertrophies, and the
epiphyseal marrow becomes necrotic. The area revascularizes, and the necrotic bone is replaced by new bone. This process can take 18 to 24
months. There is a critical point in these dual processes when the subchondral area becomes weak enough that fracture of the epiphysis
occurs. At this time, the child becomes symptomatic. With fracturing, further reabsorption and replacement by fibrous bone occurs, and the
shape of the femoral head is altered. Articulation of the head in the hip joint is interrupted. The bone re-ossifies with or without treatment; but
without treatment, the femoral head flattens and enlarges, causing joint deformity. Lateral subluxation of the femoral head is associated with
poor outcomes.
Etiology is unclear, but certain risk factors have been identified in children. These include gender, socioeconomic group, and the presence
of an inguinal hernia and genitourinary tract anomalies. Boys are affected three to five times more often than girls; incidence increases in
lower socioeconomic groups and in children with low birth weights. The disease is bilateral in 10% to 20% of children. It affects children 4 to
8 years old.
Clinical Findings
History.
There can be an acute or chronic onset with or without a history of trauma to the hip, such as jumping from a high place.
• The most common presenting sign is an intermittent limp (abductor lurch), especially after exertion, with mild or intermittent pain.
• The most frequent complaint is persistent pain in the groin, anterior hip region, or laterally around the greater trochanter.
1063
• Pain may be referred to the medial aspect of the ipsilateral knee or to the anterior thigh.
• Some children may report limited range of motion of the affected extremity.
Physical Examination.
Findings may include the following:
• Antalgic gait with limited hip movement
• Trendelenburg gait resulting from pain in the gluteus medius muscle
• Muscle spasm
• Atrophy of gluteus, quadriceps, and hamstring muscles
• Decreased abduction, internal rotation, and extension of the hip
• Adduction flexion contracture
• Pain on rolling the leg internally
Diagnostic Studies.
Routine AP pelvis and frog-leg lateral views are used to confirm the diagnosis, stage the disease, and follow disease progression
and response to treatment. Radiographic findings can include smaller epiphysis, increased epiphyseal density, subchondral
fracture line, lateralization of the femoral head, and other features. Changes in the epiphysis margin are discerned by the
orthopedist and radiologist (Fig. 38-10). However, there may be no radiographic findings early in LCPD. Ultrasonography is
useful in the preliminary diagnosis; capsular distention can be seen on sonographic images. Bone scans and MRI allow for
precise localization of the bone involvement, but changes seen as bone marrow edema and joint effusions are nonspecific. CT is
not typically used on a routine basis to evaluate patients with LCPD (Kim and Herring, 2014).
Differential Diagnosis
Acute and chronic infections, sickle cell disease, toxic synovitis, Gaucher disease, slipped capital femoral epiphysis (SCFE), osteomyelitis,
juvenile rheumatoid arthritis, hemophilia, and neoplasm are included in the differential diagnosis.
Management
• Referral to an orthopedist is necessary. Because age of onset and the severity of LCPD can vary significantly from one child to another,
there are various approaches to the management, and treatment remains controversial. Overall, the general approach is guided by the
principle of containment of the femoral head within the acetabulum. To be successful, containment must be instituted while the femoral
head is still moldable. Non-operative containment can be achieved in a variety of ways and ranges from activity limitation, and protected
weight-bearing, use of NSAID and physical therapy to maintain hip motion to bed rest with traction using casts to maintain hip abduction.
Surgical approaches involve pelvic and femoral osteotomies of the proximal femur or pelvis.
• Support and monitor the child throughout treatment and recovery, including during interruption of school or other activities. Treatment and
monitoring of LCPD can last months to years.Complications
Osteoarthritis related to femoral head deformity and decreased use of the hip joint may occur, depending on the femoral head remodeling
status. Older children have a poorer prognosis owing to the decreased opportunity for femoral head remodeling in the remaining growth
period. Females with LCPD also have a poorer prognosis.
Prevention
The condition is not preventable, but early identification and treatment reduce the long-term complications of the disorder, such as premature
degenerative arthritis in early adult life.
Idiopathic Scoliosis
Adolescent scoliosis can resolve, remain static, or increase. As a result treatment options vary considerably. Treatment decisions
are based on the natural history of each curvature. Infantile scoliosis can resolve spontaneously; however, progressive curves
require bracing and surgery in an attempt to slow the curve progression and prevent complications (e.g., thoracic insufficiency
syndrome). Juvenile scoliosis is found more frequently in girls, and the curves are at high risk for progression and often require
surgical intervention. The goal in treatment is to delay spinal fusion, allowing time for the pulmonary system and thoracic cage
to have matured and maximum trunk height to be achieved. (See the various surgical procedures described in the following
section.) The natural history includes the degree of skeletal maturity or growth remaining, the magnitude of the curve, and any
associated diagnoses or medical conditions.
Observation is always indicated for curves less than 20 degrees. Bracing or surgery may be indicated for larger curves. Brace
treatment may reduce the need for surgery, restore the sagittal profile, and change vertebral rotation. Indications for bracing are
a curve more than 30 degrees. Additional indications for brace therapy include skeletally immature patients with curves of 20 to
25 degrees that have shown more than 5 degrees of progression. The efficacy of bracing for adolescent idiopathic scoliosis
remains controversial. Some studies show brace treatment to be effective in preventing curvature progression; however, it has
been found that the success of the treatment is proportional to the amount of time that the patient wears the brace. Various brace
treatment protocols suggest wearing a brace as much as 23 hours per day; therefore, compliance is a significant factor for this
treatment modality (Spiegel and Dormans, 2011).
Surgical treatment is indicated for children and adolescents who have progressive spinal deformity that do not respond to
bracing and for those with curvature exceeding 45 to 50 degrees (Richards et al, 2014). There are various surgical procedures; all
aim to control progressive curvatures. In the past, surgery was limited to arthrodesis (surgical fusion) of the spine.
In recent years, several procedures have been developed that are designed to postpone and, in some cases, eliminate the need
for early spinal fusion and allow for growth. These include the vertical expandable prosthetic titanium rib (VEPTR). This
procedure is indicated for children with restricted pulmonary function due to the curvature of their thoracic spine. The surgery
involves implanting a prosthesis that serves to enlarge the constricted thorax. The prosthesis can be adjusted approximately
every 4 to 6 months, thereby allowing for growth. The “growing rod” surgical procedure has shown success in patients with
adolescent idiopathic scoliosis and involves inserting spinal rods that are used to exert distraction forces that are adjusted
approximately every 6 months. The rods serve as an internal brace to control the curvature of the spine while allowing skeletal
growth. A more recent procedure involves intervertebral spinal stapling or tethering. Unlike the VEPTR and growing rod
procedures, intervertebral spinal stapling does not require repeat adjustments and, therefore, eliminates the need for repeat
surgical procedures. Research on this technique is limited, and clinical indications have not been universally agreed upon.
Further research is necessary and long-term results are yet to be determined.
Referral to an orthopedist or a center that specializes in working with infants and children with scoliosis is essential.
Support must be given to the child and family 1060through the diagnostic and treatment phases, considering school and peer
factors. The primary care provider needs to assist the child with psychological adjustment issues that arise if bracing or surgery
is recommended and instituted. Some specific concerns of the child can include self-esteem problems, managing hostility and
anger, learning about the disease and its care, wondering about the long-term prognosis, and concerns about clothing and
participation in sports and other activities.
Complications
Progressive scoliosis can result in a severe deformity of the spinal column. Severe deformities can result in impairment of respiratory and
cardiovascular function and limitation of physical activities and decreased comfort. The psychological consequences of an untreated scoliosis
deformity can be severe.
Prevention
Prevention is not possible; however, screening and early identification of children with scoliosis may help avoid more expensive, invasive
care and prevent the long-term consequences of the disorder. Screening is effective, however, only if identified children are referred for care.
Parents must be notified, a referral arranged, and follow-up ensured.Scoliosis
Scoliosis is a three-dimensional deformity most commonly described as a lateral curvature of the spine in the frontal plane.
There are two types of scoliosis: nonstructural and structural. Nonstructural, also known as functional scoliosis, involves a curve
in the spine without rotation of the vertebrae. The curve is reversible, considering it is caused by conditions such as poor
posture, muscle spasms, pain, or leg length discrepancy. Structural scoliosis involves a rotational element of the spine and has
various classifications depending on the cause. The remaining discussion pertains to structural scoliosis.
The diagnosis is based on a curvature of more than 10 degrees using the Cobb method in which the angle between the
superior and inferior end vertebrae (tilted into the curve) is measured by a radiologist (see Diagnostic Studies in the following
text). In most pediatric cases, the etiology is unknown and is therefore termed and classified as idiopathic. Other classifications
include congenital, in which vertebrae fail to form (e.g., hemivertebrae), and neuromuscular (e.g., cerebral palsy,
neurofibromatosis, Marfan syndrome). Kyphosis, which results from disorders of sagittal alignment (such as, postural kyphosis
and Scheuermann disease), is another classification of structural scoliosis. Kyphosis, commonly termed round back, is discussed
following scoliosis.
• Idiopathic: Etiology is unknown and is likely multifactorial. It is the most common type of scoliosis. There are three types
classified by age at onset:
• Infantile (0 to 3 years old)
• Juvenile (3 to 10 years old)
• Adolescent (11 years old and older)
• Congenital: A structural anomaly present at birth (e.g., hemivertebrae) often associated with other congenital abnormalities,
such as renal and cardiac anomalies; progression of curvature can worsen rapidly, particularly during periods of rapid growth
(e.g., first 2 to 3 years of life and adolescence).
• Neuromuscular: Most common in non-ambulatory patients. Secondary to weakness/imbalance/spasticity of the muscles of the
trunk caused by primary neuromuscular problems (e.g., cerebral palsy or muscular dystrophy). In contrast to idiopathic and
congenital scoliosis, curves caused by neuromuscular disorders can continue to progress after skeletal maturity.
Idiopathic is the most common type of scoliosis. Its etiology is unknown, but it often has a familial or genetic pattern. The
overall incidence of idiopathic scoliosis is approximately 2% to 3% with between 0.3% and 0.5% of children with scoliosis having
curves greater than 20 degrees on radiography and less than 0.1% demonstrating curves greater than 40 degrees Cobb's angle.
Hormonal changes play a role in the disease process, and a rapid growth period is believed to be a significant factor in the
progression of curvature associated with idiopathic scoliosis. In addition, the risk of curve progression depends on the amount
of growth remaining, the magnitude of the curve, and gender. Although the incidence of idiopathic scoliosis is nearly equal in
girls and boys, females have a much higher risk of developing curves more than 30 degrees (Spiegel and Dormans, 2011). The
most common type of idiopathic scoliosis is found in adolescents and is the major focus of the remaining discussion.
Small to moderate scoliotic curves (10% to 30%) usually do not increase significantly after skeletal growth is complete but
bear watching, particularly during periods of rapid growth velocity. Double S-curves and more severe curves are more likely to
progress during the growth years. For a given child, however, the ability to predict progression is difficult, because even small
curves (10% to 25%) can progress to severe deformity. Thus regular monitoring of any curve in a skeletally immature child is
important (Table 38-2).
The female-to-male ratio increases with increasing curve magnitude. For curves less than 20 degrees, the risk for progression of the curve
is low; these curves generally just need to be observed. However, for curves between 20 and 45 degrees, the risk for progression is high
during growth, and early intervention is of paramount importance. In children with curves greater than 50 degrees, the spine loses its ability
to compensate and progression is expected. Young premenarchal females with large curves are a vulnerable group, because their spines are
skeletally immature with growth remaining. The majority of adolescents with idiopathic scoliosis have a right thoracic curve. Juvenile
manifestation is uncommon, and infantile scoliosis is rare in the United States.
Clinical Findings
History.
Scoliosis is generally painless, and insidious onset is typical. Generally there is no significant history. The provider should assess the
following:
• Family history of scoliosis
• Age of menarche
• Etiologic factors related to the various causes of structural scoliosis
The presence of pain with a lateral curvature of the spine suggests an inflammatory or neoplastic lesion as the cause of the scoliosis. Some
children with idiopathic scoliosis complain of mild pain that is activity related. Severe, constant, or night pain and point tenderness could be
indicative of other pathologic conditions (e.g., metastatic tumor or stenosis) and warrants further investigation.Physical Examination.
Children of all ages should be evaluated in the standing position, from both the front and the side, to identify any asymmetry. Looking
primarily at the straightness of the spine can be misleading, because scoliosis involves both rotation and misalignment of the vertebrae. The
Adams forward bend position accentuates 1058the rotational deformity of scoliosis. Asymmetries to look for include:
• Unequal shoulder height
• Unequal scapula prominences and heights: Note that the muscle masses may be somewhat unequal, especially if the child uses one shoulder
more than the other as in carrying books. Look for bony, not muscular, prominence.
• Unequal waist angles: The hip touches one arm, and the contralateral arm hangs free.
• Unequal rib prominences and chest asymmetry
• Asymmetry of the elbow to flank distance, and some deviation of the spine from a straight head-to-toe line
• Unequal rib heights when the child stands in the Adams forward bend position (see Fig. 38-8)
During the Adams test the examiner looks for asymmetry of the posterior chest wall on forward bending, the earliest abnormality seen.
Rotation of the vertebral bodies toward the convexity results in outward rotation and prominence of the attached ribs posteriorly. The anterior
chest wall may be flattened on the concavity due to inward rotation of the chest wall and ribs. Associated findings may include elevation of
the shoulder, lateral shift of the trunk, and an apparent leg-length discrepancy.
Congenital scoliosis may be visible in the infant lying prone; it is sometimes more prominent if the infant is suspended prone. Inspect for
skin abnormalities, sacral dimple, and hairy patches.
The physical examination should also include the following:
• Observation for equal leg lengths
• Examination of the skin for hairy patches, nevi, café au lait spots, lipomas, dimples
• Neurologic examination checking for weakness or sensory disturbance
• Cardiac examination with diagnosis of Marfan syndrome
Diagnostic Studies.
Standing AP and lateral radiographs of the entire spine are recommended at the initial evaluation for patients with clinical findings suggestive
of a spinal deformity. On the PA radiographs, the degree of curvature is determined by the Cobb method. An MRI is helpful when an
underlying cause for the scoliosis is suspected based on age (infantile, juvenile curves), abnormal findings in the history and on physical
examination, and atypical radiographic features. Atypical radiographic findings include uncommon curve patterns, such as the left thoracic
curve, double thoracic curves, high thoracic 1059curves, widening of the spinal canal, and erosive or dysplastic changes in the vertebral body
or ribs. On the lateral radiograph, an increase in thoracic kyphosis or an absence of segmental lordosis may be suggestive of an underlying
neurologic abnormality (Spiegel and Dormans, 2011).
Differential Diagnosis
Structural scoliosis must be differentiated from functional scoliosis. The latter disappears when the child is placed in the Adams forward bend
position, whereas the former is enhanced in this position. Persistent functional scoliosis to one side in a child with a neuromotor problem can
eventually become structural and must be managed with physical therapy or other means to prevent progression. Consider systemic problems,
such as neurofibromatosis, cerebral palsy, multiple sclerosis, Rett syndrome, rickets, tuberculosis, and tumor.
Management
The primary aim of scoliosis management is to stop curvature progression and improve pulmonary function. Treatment options include
observation, bracing, and surgical treatment. The management presented in this text addresses idiopathic scoliosis treatment. Of note, genetic
testing is now available to provide a personalized treatment approach for selected patients diagnosed with adolescent idiopathic scoliosis. The
ScoliScore is a genetic test that screens for more than 50 genetic markers (53 single nucleotide polymorphisms [SNPs]) linked to the
progression of spinal curves and assigns a quantitative score to a patient's deoxyribonucleic acid (DNA) saliva sample. (See the SNP
discussion in Chapter 41.) The score identifies the patient as having a low, medium, or high risk for curve progression (Bohl et al,
2014; Smith and Cruz, 2010). For children in the low-risk group, these prognostic data may lead to less frequent follow-up visits to
specialists, avoiding or discontinuing bracing, and fewer radiologic tests. However, the test is only indicated for children with mild curves (10
to 25 degrees) with growth remaining (Ward et al, 2010). Combined with diagnostic information and clinical judgment, the results of the
ScoliScore test serve as a guide for health care providers to optimize the treatment of scoliosis.
Osgood-Schlatter Disease
Osgood-Schlatter disease is caused by microtrauma in the deep fibers of the patellar tendon at its insertion on the tibial tuberosity. The
diagnosis is usually based on history and physical examination. The quadriceps femoris muscle inserts on a relatively small area of the tibial
tuberosity. Naturally high tension exists at the insertion site. In children, additional stress is placed on the cartilaginous site as a result of
vigorous physical activity, leading to traumatic changes at insertion.
Osgood-Schlatter disease is often seen in the adolescent years after undergoing a rapid growth spurt the previous year. It occurs more
frequently in boys than girls, with a male-to-female ratio of 3 : 1. This difference is probably related to a greater participation in specific risk
activities by boys than by girls (Sullivani, 2015).Clinical Findings
History.
• Recent physical activity (such as, running track, playing soccer or football, or surfboarding) commonly produces the condition.
• Pain increases during and immediately after the activity and decreases when the activity is stopped for a while.
• Running, jumping, kneeling, squatting, and ascending/descending stairs exacerbate the pain.
• The pain is bilateral in 20% to 50% of cases.
• Approximately 25% of patients give a history of precipitating trauma.
Physical Examination.
Characteristic findings include the following (Sullivan, 2015):
• Pain may be reproduced by extending the knee against resistance, stressing the quadriceps, or squatting with the knee in full flexion
• Focal swelling, heat, and point tenderness at the tibial tuberosity
• Full range of motion of knee
Diagnostic Studies.
The diagnosis is based on history and physical examination. Radiographs are not needed unless another pathologic condition is suspected.
Differential Diagnosis
Other knee derangements, tumors (osteosarcoma), and hip problems with referred pain should be considered. The referred pain of hip
problems is diffuse across the distal femur without point tenderness at the tibial tubercle.
Management
Osgood-Schlatter disease is a self-limiting condition, with symptom management the key consideration. The following steps are taken:
• Avoid or modify activities that cause pain until the inflammation subsides.
• Ice or cold therapy to reduce pain and inflammation.
• Once the acute symptoms have subsided, quadriceps-stretching exercises, including hip extension for complete stretch of the extensor
mechanism, may be performed to reduce tension on the tibial tubercle. Stretching of the hamstrings may also be useful.
• Use of NSAIDs is recommended by some but thought ineffective by others. Because this condition may last up to 2 years, their chronic use
may be problematic.
• A neoprene sleeve over the knee may help stabilize the patella.
• A patella tendon strap that wraps around the joint just below the knee reduces the strain on the tibial tuberosity.
• Cylinder casting or bracing with limited weight bearing for 2 to 3 weeks may be used in severe cases.
Complications
In the postpubertal child, a residual ossicle in the tendon next to the bone may cause persistent pain. Surgical removal is indicated and will
relieve the pain.
Prevention
The condition cannot be prevented, but earlier management may decrease the length of disability and the discomfort 1069associated with it.
Avoid overuse and encourage balanced training and adequate warm-up before exercise or sports participation. The use of kneepads may help
protect the tibial tuberosity from direct injury for those who engage in sports that result in knee contact.
Febrile Seizures
Febrile seizures are the most common type of seizures in children. They are brief, generalized, clonic or tonic-clonic in nature, and can be
either simple or complex. A concurrent illness is present with rapid fever rise to at least more than 102.2° F (39° C), but the fever is not
necessarily that high at the time of the seizure. It is conjectured that these seizures may be related to peak temperature reached during the
febrile episode. Minimal postictal confusion is associated with febrile seizures. Simple febrile seizures last less than 15 minutes and may
recur during the same febrile illness period. Complex febrile seizures last longer than 15 minutes, can recur on the same day, and can have
focal attributes (even during the postictal phase). Febrile SE is uncommon, rarely stops spontaneously, is fairly resistant to medications, and
can persist for a long period of time. Most children in febrile SE require one or more medications to end the seizure. A report found that
reducing the time from seizure onset to anticonvulsant medication administration was key to reducing the seizure duration during an episode
(Seinfeld et al, 2014).
The etiology of febrile seizures is unclear and by definition excludes seizures that are caused by intracranial illness or are related to an
underlying CNS problem. The risk is higher in children with a family medical history for febrile seizures or in those with predisposing factors
(e.g., neonatal intensive care unit [NICU] stay more than 30 days, developmental delay, day care attendance).The age range associated with febrile seizures is 6 months to 60 months. Male gender is a minor risk factor as is a lower sodium level.
Approximately 2% to 5% of neurologically healthy infants and young children experience at least one simple febrile seizure with about 30%
of this group experiencing a second episode (Mikati, 2011).
Clinical Findings
History.
Include the following:
• Description of seizure duration, type (generalized or focal), frequency in 24 hours
• Relationship of the seizure to a febrile episode and level of temperature
• Any abnormal neurologic findings noted before the seizure (is not consistent with a febrile seizure)
• Family history of afebrile or febrile seizures
• Maternal smoking in the perinatal period
• Prematurity or neonatal hospitalizations for more than 28 days
• Parents' perception of development of child
Physical Examination.
The physical examination is the same as that described earlier for seizures.
Diagnostic Studies.
Diagnostic studies include the following:
• A lumbar puncture may be done in infants younger than 12 months old and who may also have used an antibiotic prior to seizure onset,
and/or in those who have signs of meningeal irritation.
• Blood glucose in all children.
• CBC, calcium, electrolytes, and urinalysis are optional but frequently included.
• EEG if neurologic signs are present or seizure was atypical.
• MRI for complex febrile seizure features or if any doubt exists about the diagnosis.
Differential Diagnosis
Consider sepsis, meningitis, metabolic or toxic encephalopathies, hypoglycemia, anoxia, trauma, tumor, and hemorrhage. Febrile delirium
and febrile shivering can be confused with seizures. Breath-holding spells can mimic febrile seizures; however, the former are always related
to crying or tantrums. Febrile seizures come at unpredictable times during sleep, eating, play, or other generally calm times and are related to
the onset of an illness. Epileptic seizures occur without concurrent illness and at unpredictable times.
Management
• Protect the airway, breathing, and circulation if the seizure is still occurring. Place the child in a side-lying position to prevent aspiration or
airway obstruction.
• Do not put anything into the child's mouth during the seizure.
• Time the duration of the seizure and observe whether it is focal or generalized.
• Reduce the fever with acetaminophen or ibuprofen (oral or suppository) after the seizure has stopped, although the use of antipyretics will
not necessarily prevent another febrile seizure.
• The child should be seen shortly after the seizure. Advise transport to an emergency center if the seizure lasts more than 10 minutes.
• Most medical providers agree that anticonvulsants are not recommended for febrile seizures, but they may be considered if the child has
abnormal neurologic findings or developmental delays; the initial seizure was complex febrile, and there is a family history of afebrile
seizures; 683or if the child has recurrent, prolonged simple febrile seizures.
Prophylaxis for Recurrent Febrile Seizures
Prolonged anticonvulsant prophylaxis is not recommended. In the rare instance that prophylaxis is indicated, diazepam by mouth 0.33 mg/kg
every 8 hours (1 mg/kg/24 hours) can be given over the course of the febrile illness (usually for 2 to 3 days). Another approach is to use rectal
diazepam in a gel form (dosed at 0.5 mg/kg for children 2 to 5 years of age) at the time of a seizure; this will prevent recurrence for
approximately 12 hours. Side effects of diazepam include transient ataxia, lethargy, and irritability that can be decreased by adjusting the
dosage.
Antipyretics can reduce the discomfort associated with a fever but do not alter the risk of having another febrile seizure. The thought as to
why antipyretics are not helpful as prophylactic agents involves the mechanism implicated in a simple febrile seizure which likely takes place
when the temperature is either rising or falling (Mikati, 2011).Education
The family should receive information about febrile seizures, their risks, and their management. Education should include information
explaining the febrile seizure, reassurance that no long-term consequences are associated with febrile seizures, information that febrile
seizures recur in some children and that nothing can be done to prevent the seizures, and first-aid information in case another seizure occurs
at some time. The decision to use prophylaxis is up to the parents and the PCP on a case-by-case basis. A follow-up phone call after the event
is useful.
Complications
Death or persisting motor deficits do not occur in patients with febrile seizures. No indication has been found that intellect or learning is
impaired. An affected child has an increased risk for the development of epilepsy (less than 5%) if the seizure is prolonged and focal; if the
child has repeated seizures with the same febrile episode; or if the child has had a prior neurologic deficit, a family history of epilepsy, or
both. Two thirds of children who have had one simple febrile seizure will have no more. The younger the age at onset (younger than 18
months old) of the first febrile seizure, the lower the temperature threshold that is needed to cause the child to seize and the more likely the
child is to have a recurrence.
Testicular Torsion
Testicular torsion is the result of twisting of the spermatic cord, which subsequently compromises the blood supply to the testicle. Generally,
there is a 6-hour window following a testicular torsion before significant ischemic damage and alteration in spermatic morphology and
formation occurs (Elder, 2011b).
Normal fixation of the testis is absent, so the testis can rotate and block lymphatic and then blood flow. Torsion can occur after physical
exertion, trauma, or on arising. Torsion can occur at any age but is most common in adolescence and is uncommon before 10 years old. The
left side 946is twice as likely to be involved because of the longer spermatic cord.
Clinical Findings
History
• Sudden onset of unilateral scrotal pain, often associated with nausea and vomiting. The pain is unrelenting.
• History of bouts of intermittent testicular pain. Prior episodes of transient pain are reported in about half of patients.
• Minor trauma, physical exertion, or onset of acute pain on arising is possible.
• May be described as abdominal or inguinal pain by the embarrassed child.
• Fever is minimal or absent.
Physical Examination
• Ill-appearing and anxious male, resisting movement
• Gradual, progressive swelling of involved scrotum with redness, warmth, and tenderness
• The ipsilateral scrotum can be edematous, erythematous, and warm
• Testis swollen larger than opposite side, elevated, lying transversely, exquisitely painful
• Spermatic cord thickened, twisted, and tender
• Slight elevation of the testis increases pain (in epididymitis it relieves pain)
• Transillumination can reveal a solid mass
• The cremasteric reflex is absent on the side with torsion
• Neonate—hard, painless, non-transilluminating mass with edema or discolored scrotal skin
Diagnostic Studies
• UA is usually normal and pyuria and bacteriuria indicate UTI, epididymitis, or orchitis.
• Doppler ultrasound: Testicular flow scan considered if Doppler ultrasound within normal and time allows.
Differential Diagnosis
Torsion of the testicular or epididymal appendage, acute epididymitis (mild to moderate pain of gradual onset), orchitis, trauma (pain is better
within an hour), hernia, hydrocele, and varicocele are included in the differential diagnosis.
Management
Testicular torsion is a surgical emergency, and identification with prompt surgical referral must occur immediately. Occasionally manual
reduction can be performed, but surgery should follow within 6 to 12 hours to prevent retorsion, preserve fertility, and prevent abscess and
atrophy. Contralateral orchiopexy may be done because of a 50% occurrence of torsion in nonfixed testes. Rest and scrotal support do not
provide relief.Patient and Family Education, Prevention, and Prognosis
Testicular atrophy, abscess, or decreased fertility and loss of the testis as a result of necrosis can occur if the torsion persists more than 24
hours.
Disease
Clinical Presentation
Laboratory Diagnosis Treatment
History Physical Findings
Iron deficiency Fatigue
Irritability
Excess milk
intake
Pallor or none RBC hypochromic, microcytic
MCV ↓
Serum iron ↓
TIBC ↑
Percentage of saturation
↓
Ferritin ↓
Blood in stool or urine
Ratio of MCV/RBC >13
Correct diet
Eliminate source of bleeding
Ferrous SO4 up to 6 mg/kg/day of
elemental iron
Iron Deficiency Anemia
IDA is the most common nutritional disorder and hematologic condition in the world. Approximately 3% to 7% of children at
age 1 year suffer from iron deficiency (Powers and Buchanan, 2014). Nine percent of adolescent girls develop iron deficiency
and 2% to 3% between the ages of 12 and 19 years old develop anemia primarily due to rapid growth, heavy menses, and
nutritionally inadequate diets 641(Abrams, 2014). The incidence of IDA among children in the United States has been declining
slightly during the past four decades, although the prevalence remains high among children living at or below poverty level and
in black and Hispanic children. Other risk factors include childhood obesity and a history of prematurity or low birth weight
(Mahoney, 2015). Iron deficiency correlates with rapid increases in body size and blood volume during the first 2 years, along
with diets low in iron, such as occurs with an overuse of goat's or cow's milk. The deficient iron intake is also associated with
prolonged bottle-feeding.
Dietary iron is absorbed throughout the intestine but especially in the duodenum. Malabsorption of iron occurs in diseases
that affect this segment of the intestine, such as celiac disease, Crohn disease, giardiasis, or resection of the proximal small
intestine. Disorders causing GI blood loss such as inflammatory bowel disease, cow's milk–induced colitis, or chronic use of
aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs) also deplete iron stores and contribute to iron deficiency (Mahoney,
2015).
Anemia (Hgb level <11 g/dL) is neither a sensitive nor a specific screen for iron deficiency because about two thirds of irondeficient children are not anemic. Conversely, the detection of anemia is not specific to iron deficiency, because two thirds of
anemic children have another cause for their anemia. The minimum laboratory screening for iron deficiency is the Hgb level.
Often, the simplest and most cost-effective measurement is a CBC, which includes the Hgb, Hct, MCV, and RDW. A ferritin
level may also be helpful because this indicates body stores of iron, but it must be interpreted carefully because ferritin is an
acute phase reactant and may be increased with inflammatory conditions (Mahoney, 2015). The American Academy of
Pediatrics (AAP) Committee on Nutrition recommends universal Hgb screening for anemia at 12 months old (Baker and Greer,
2010). This screening should include an assessment of risk factors for iron deficiency and IDA (Box 27-2).
Screening Hgb can be performed on children younger than 1 year old when risk factors warrant it. Menstruating females may
also require screening for IDA due to the monthly blood loss, rapid growth, and potentially inadequate diet. When screening for
IDA or any other routine health screening recommendation, remember that screening is not just a one-time test; the effectiveness
of treatment must be determined through follow-up testing. Thus after the routine 12-month Hgb/Hct testing, risk assessment
for anemia should be performed at all preventive pediatric health care visits with follow-up blood testing if positive. If children
are at risk for IDA, a repeat Hgb/Hct should be performed as often as indicated.
Wilms TumorWilms tumor, the most common malignancy of the genitourinary tract, is typically recognized as a firm, smooth 937mass in the
abdomen or flank. It is staged according to the Children's Oncology Group as follows:
• Stage I: The tumor is limited to the kidney and can be completely excised with the capsular surface intact.
• Stage II: The tumor extends beyond the kidney but can still be completely excised.
• Stage III: There is postsurgical residual nonhematogenous extension confined to the abdomen.
• Stage IV: There is hematogenous metastasis, most frequently to the lung.
• Stage V: There is bilateral kidney involvement.
This malignancy manifests as a solitary growth in any part of either or both kidneys. There are approximately eight cases of
Wilms tumor per million children younger than 15 years old with 500 new cases every year. Most Wilms tumors occur in
children between 2 and 5 years old. The peak incidence and median age at diagnosis is 3 years old. About 1% to 2% of children
with Wilms tumor have a family history of Wilms, and the tumor is inherited in an autosomal dominant manner (Anderson
et al, 2011). An important feature of Wilms tumor is the occurrence of associated congenital anomalies including renal
abnormalities, such as cryptorchidism, hypospadias, duplication of the collecting system, ambiguous genitalia,
hemihypertrophy, aniridia, cardiac abnormalities, and Beckwith-Wiedemann, Denys-Drash, and Perlman syndromes. Wilms
tumor occurs with equal frequency in both sexes although males are diagnosed younger. There is a higher frequency in African
Americans and a lower frequency in Asians.
Clinical Findings
History
• The most frequent finding is increasing abdominal size or an actual palpable mass.
• Pain is reported if the mass has undergone rapid growth or hemorrhage.
• Fever, dyspnea, diarrhea, vomiting, weight loss, or malaise may be reported.
Physical Examination
• A firm, smooth abdominal or flank mass that does not cross the midline may be noted.
• BP is elevated if renal ischemia is present (rare).
• A left varicocele is found in males if the spermatic vein is obstructed.
• A careful examination is needed to rule out congenital anomalies.
Diagnostic Studies
• Chest and abdominal radiography is performed to differentiate neuroblastoma, which is usually calcified.
• Abdominal ultrasonography is used to differentiate a solid from a cystic mass or hydronephrosis and multicystic kidney.
• UA demonstrates hematuria in 25% to 33% of children.
• A CBC, reticulocyte count, and liver and renal chemistry studies are performed.
• A CT scan of the chest, abdomen, and pelvis to stage the disease and bone marrow is done by the oncology team.
Differential Diagnosis
Neuroblastoma is the main differential diagnosis (the mass often crosses the midline). Multicystic kidney, hydronephrosis, renal cyst, or other
renal malignancies are additional conditions to consider.
Management
Diagnostic workup is the initial urgent priority, with concurrent referral to a pediatric cancer center for treatment. Surgery is scheduled to
remove the affected kidney and possibly the ureter and adrenal gland; combined chemotherapy and radiotherapy are instituted if the disease is
advanced or histologic findings are unfavorable. Close follow-up after the initial treatment should be coordinated with the cancer team.
Complications
The lungs and liver are the most common sites of metastasis. High BP is possible because of renal ischemia and occasionally leads to cardiac
failure. Scoliosis resulting from radiation therapy is uncommon because radiation exposure is carefully controlled.
Patient and Family Education, Prevention, and Prognosis
The prognosis is determined by the histology of the neoplasm, by the patient's age (the younger the better), the size of the tumor, positive
nodes, and, most significantly, the extent or stage of the disease. The cure rate is about 80% to 90% for infants with stage 4S; children with
high-risk neuroblastoma have survival rates between 25% to 35%; reoccurrence of the disease has a less than 50% response to alternativechemotherapeutic agents (Zage and Ater, 2011). A pediatric urologist should determine if a child should be allowed to participate in sports on
an individual basis (American Academy of Family Physicians [AAFP] et al, 2010). Use of kidney protectors is highly recommended
during sports. New information on the long-term sequelae for the treatment of Wilms tumors and the present trials and treatment
recommendations can be accessed at the National Wilms Tumor Study
Growth Disorders (Turner’s Syndrome)
Children grow in a predictable way, and deviation from a normal growth pattern can be the first sign of an endocrine disorder.
Accurate serial growth data must be collected in order to assess a pattern of growth and current growth velocity. A child's
predicted growth potential is based in large part on genetic potential and may change with altered nutritional status and illness
patterns. An estimate of the expected stature (±2 standard deviations where 1 standard deviation equals 2 inches [4.5 cm]) for a
particular child can be made by calculating a mid-parental target height:
• Target height for boys: (Mother's height + 5 inches [13 cm]) + (Father's height)/2
• Target height for girls: (Father's height − 5 inches *13 cm]) + (Mother's height)/2
Growth disorders may be classified as primary or secondary. Primary growth disorders include skeletal dysplasias,
chromosomal abnormalities (e.g., Turner syndrome), and genetic short stature. Secondary growth disorders may result from
undernutrition, chronic disease, endocrine disorder, and idiopathic (constitutional) growth delay (CGD) (Box 26-1). The
following discussion focuses on growth hormone deficiency (GHD) and CGD (Table 26-1).
Karyotype to rule out Turner syndrome in girls: Girls with Turner mosaicism may not manifest the typical clinical findings
of Turner syndrome (e.g., cubitus valgus, webbing of the neck), thus highlighting the importance of karyotyping all females
presenting with short stature (Milbrandt and Thomas, 2013) (see Chapter 41).
Early Puberty/Precocious Puberty
Early puberty is divided into four categories: premature thelarche, premature adrenarche, isolated menarche, and true
precocious puberty.
Premature thelarche, isolated breast development without any other features of puberty, occurs in infant and toddler girls
and is sometimes present at birth. This breast development, likely due to estrogens produced during the mini puberty of infancy
or increased responsiveness of the breast primordia, resolves over time and rarely progresses to true precocious puberty.
Premature adrenarche is the early onset of pubic or axillary hair in either boys (prior to 10 years old) or girls (prior to 8 years
old) not associated with other features of true puberty. Bone and height age may be slightly advanced in relation to chronologic
age in children with premature adrenarche and plasma dehydroepiandrosterone (DHEA) values may be slightly elevated
(Loomba-Albrecht and Styne, 2012). Premature adrenarche may be caused by a mild form of congenital adrenal hyperplasia
(CAH), exposure to topical testosterone, or rarely, adrenal tumor. Most often, the condition is idiopathic. Children with
idiopathic premature adrenarche are at increased risk for polycystic ovary syndrome and metabolic syndrome (Bordini and
Rosenfield, 2011b).
Isolated menarche is an uncommon condition in which girls have one to a few episodes of vaginal bleeding without breast
development. In this condition, sexual abuse, vaginal tumor, a functional estrogen-producing ovarian cyst, and primary
hypothyroidism all need to be excluded.
602
True precocious puberty refers to the onset of multiple features of puberty earlier than the normal range. It is defined as
thelarche or pubarche (appearance of pubic hair) before 8 years old in girls and before 9 years old in boys, except in the case of
non-Hispanic African American and Mexican American girls where thelarche is considered within the normal range after 7
years old (Bordini and Rosenfield, 2011b). Features of precocious puberty may include accelerated linear growth, breast
development or penile enlargement, and pubic hair development. Depending on the duration of symptoms, the bone age may
be advanced. Precocious puberty can be divided into two broad categories: (1) central, gonadotropin dependent; or (2)
peripheral, gonadotropin independent (Box 26-4). Prolonged exposure to exogenous sex hormones (mother's birth control pills
or father's topical testosterone) (Rodriquez and Dougan, 2013) and exposure to chemicals that disrupt endocrine function
(see Chapter 42) can cause precocious puberty (Ozen and Darcan, 2011).
In the United States, the incidence of precocious puberty is 0.01% to 0.05% per year. Precocious puberty is more common in females
compared to males and in African American children compared to Caucasian children (Rodriquez and Dougan, 2013). Any lesion that
disrupts the normal connections between the brain and the hypothalamus can cause central precocious puberty. This condition is most often
idiopathic in girls. Boys have a 30% incidence of CNS tumors in situations of central precocious puberty.
Clinical Findings
Many children who present with features of early puberty do not require treatment. All children who exhibit signs of puberty at a younger age
than normal, however, should have an evaluation as to the etiology. Those children who start to develop signs of puberty at the early end ofthe normal range should be evaluated if they have rapid progression of pubertal signs resulting in a bone age more than 2 years ahead of
chronologic age, or new CNS-related findings (e.g., headaches, seizures, and/or focal neurologic defects).
History.
Evaluation includes the following:
• Age of onset
• Type, duration, and progression of pubertal symptoms (i.e., breast tissue, pubic hair, phallic enlargement, acne, body odor, oily scalp)
• Pattern of growth
• Any symptoms suggestive of a CNS lesion
• Family pattern of pubertal changes
• Exposure to topical estrogens or testosterone, oral estrogens, or environmental hormone disruptors
Physical Examination.
Physical examination should include:
• Assessment of stature and growth velocity
• Description of the child's Tanner stage:
• Breast development: Breast development should be evaluated by palpation rather than inspection to differentiate between the presence
of true breast tissue versus fat deposition
• Presence of pubic and axillary hair (girls)
• Penile length, testicular volume, and pubic and axillary hair (boys) (see Chapter 8)
Diagnostic Studies.
Diagnostic studies should include:
• Premature thelarche: No laboratory studies are necessary in the infant or toddler girl unless she has other features of true puberty or
continued increase in breast size.
• Premature adrenarche: Serum 17-hydroxyprogesterone (17-OHP) to exclude CAH and a 24-hour urine collection for 17-ketosteroids or
imaging of the adrenal glands to exclude an adrenal tumor.
• Isolated menarche: Thyroid function tests to exclude primary hypothyroidism, and pelvic ultrasound to rule out the presence of an ovarian
cyst or pelvic tumor.
• True precocious puberty:
• Bone age x-ray of left wrist
• LH, FSH, and estradiol or testosterone: Use a laboratory with a sensitive assay that will detect early pubertal values at the lower end of
the range.
• If LH and FSH are high (in pubertal range: indication of central etiology), an MRI is indicated to exclude CNS tumor.
• If LH and FSH are low (in prepubertal range: indication of peripheral puberty), complete a GnRH stimulation test to distinguish
central from peripheral puberty.
• If etiology is peripheral puberty:
• Pelvic ultrasonography of girls
• Testicular ultrasonography of boys
• Serum 17-OHP to rule out a severe form of CAH
603
Management
Treatment of early puberty depends on the etiology and should always be done with the guidance of a pediatric endocrinologist.
Management depends on the underlying disorder, age of the child, degree of advancement of the bone age, and the child's and family's
emotional response to the condition. Radiation, surgery, or chemotherapy is indicated in the case of CNS tumors. A long-acting GnRH
agonist may be used to bring serum sex steroids to prepubertal levels. Treatment of precocious puberty is important to increase final adult
height.
Female Stages (Menarche)
Females enter puberty earlier than males do, and their puberty usually progresses sequentially in the following pattern:
• Ovaries increase in size; no visible body changes occur.
• Breast budding (thelarche) traditionally occurs between 9 and 10 years old, with 97% of girls having initial breast development by 12 years
old (Cabrera et al, 2014) (Fig. 8-3). Evidence indicates that adolescent girls are entering and completing puberty younger than girls did 50
years ago, with the average age decreasing by 1 year in the past few decades (Biro et al, 2012; Cabrera et al, 2014). Most girls (85%)
experience the development of breast buds approximately 6 months before the appearance of pubic hair. African American girls, onaverage, reach thelarche and onset of menstruation (menarche) approximately 6 months prior to their Caucasian peers (Cabrera et al, 2014).
The timing of the onset of breast development in females has no relationship to breast size at the completion of puberty.
• Rapid linear growth usually begins shortly after the onset of breast budding and reaches its peak about 1 year later. Ninety-five percent of
females reach peak height velocity (PHV) between the ages of 10 and 14 years, and most girls experience PHV about 6 to 12 months before
menarche, generally between 11 and 12 years old (Busscher et al, 2012). Early developers may experience a height 122spurt between 9 and
10 years old, whereas late developers may not experience a height spurt until between 13 and 14 years old. Final height is determined by the
amount of bone growth at the epiphyses of the long bones. Growth stops when hormonal factors shut down the epiphyseal plates.
• Appearance of pubic hair (adrenarche or pubarche) commences at about years old and is related to adrenal rather than gonadal
development, not to thelarche; therefore, it is less valid than other secondary sex characteristics in assessing sexual maturation (Fig. 8-4).
• The first menstrual period (menarche) occurs, on average, at years old. More than 95% of girls experience menarche
between and years old. The mean age of menarche is highly dependent on ethnic, socioeconomic, and nutritional factors.
Menarche generally occurs approximately years after thelarche (Cabrera et al, 2014). It may be 18 to 24 months after menarche
before females establish regular ovulatory cycles. To some degree, menstrual cycles can be affected by athletic activity. The American
Academy of Pediatrics (AAP) and the American Congress of Obstetricians and Gynecologists (AGOG) recommend that health care
providers recognize the menstrual cycle as a ―vital sign‖ because of the need for education regarding normal timing and characteristics of
menstruation and other pubertal signs (ACOG Committee on Adolescent Health Care, 2006; Hagan et al, 2008).
Changes in the body composition of females occur during puberty, and adolescent girls benefit from the primary health care provider's
reassurance that these changes are normal. Initial breast development usually begins as a unilateral disk-like subareolar swelling, and many
adolescents and parents may initially present with concerns about breast tumors. Girls often have asymmetric breasts and need 123assurance
that breasts become more or less the same size within a few years after the onset of breast budding. The female body shape changes as girls
progress through puberty, with broadening of the shoulders, hips, and thighs. Girls experience a continuous increase in proportion of fat to
total body mass during puberty. They enter puberty with approximately 80% lean body weight and 20% body fat. By the time puberty ends,
lean body mass drops to about 75%. Body fat is an important mediator for the onset of menstruation and regular ovulatory cycles. An average
of 17% of body fat is needed for menarche, and about 22% is needed to initiate and maintain regular ovulatory cycles.
Dysmenorrhea
Painful menstruation with cramping in the lower abdomen or pelvis is the most common gynecologic problem seen in adolescence. Primary
dysmenorrhea has no pelvic pathologic condition identified, whereas secondary dysmenorrhea is due to a pelvic pathologic condition.
968
Primary dysmenorrhea is painful menses caused by an exaggerated production of prostaglandins, primarily prostaglandin F2α, in the
secretory endometrium. This causes uterine contractions and vasoconstriction leading to ischemia and pain. The elevation of prostaglandins is
brought about by falling progesterone levels during the luteal phase of ovulatory cycles.
Secondary dysmenorrhea may be prompted by endometriosis; complications of pregnancy; outflow obstruction; ovarian cysts, fibroids, or
other uterine abnormalities; or infection. Dysmenorrhea is present in more than 50% of adolescents and has been reported in up to 93%. It is
the leading cause (greater than 10%) of absenteeism from school or work, with increasing incidence in those who describe the pain as severe
(Laufer, 2012).
Clinical Findings
History
For primary dysmenorrhea, ask about the following:
• Menstrual history
• Attitudes and beliefs about menstruation
• Onset—usually 6 to 24 months after menarche
• Location—lower midabdominal area radiating to back, thighs
• Duration and timing of pain—usually begins with menses and lasts less than 2 days
• Character—mild to severe cramping
• Associated symptoms—nausea, vomiting, diarrhea, headache, fatigue, nervousness, dizziness, urinary frequency, lower back or thigh pain
• Ameliorating or aggravating factors
• Treatments or medications tried, including complementary and alternative medicine (CAM)
• Sexual activity
• Number of days of school or activities missed
• Cigarette smoking• Family history of dysmenorrhea
For secondary dysmenorrhea, the following history should be further explored:
• Onset (with menarche or 2 to 3 years postmenarche)
• Pelvic pain at times other than menstruation (worsens over time)
• Character of pelvic pain (dull and constant rather than crampy)
• History of infection, menorrhagia, intermenstrual bleeding, or abnormal vaginal discharge
• Dyspareunia
• History of sexual abuse
• Family history of endometriosis
Physical Examination
A complete physical examination is recommended and required for secondary dysmenorrhea. A speculum and bimanual examination may be
deferred if the adolescent is not sexually active, if the dysmenorrhea is mild, if it does not interfere with daily activities, or if the
dysmenorrhea is responding to treatment and without suspicion of pathologic condition. However, the external genitalia should be examined
and a cotton swab inserted into the vagina to rule out hymenal abnormalities and/or a vaginal septum. A rectoabdominal examination also
helps rule out adnexal tenderness and masses (Laufer, 2012).
Diagnostic Studies
The following are ordered only if indicated:
• NAATs or cervical cultures for gonorrhea and chlamydia
• CBC with sedimentation rate if PID is suspected
• Pregnancy test
• Pelvic ultrasound if abnormalities are suspected
Differential Diagnosis
Endometriosis, PID, chronic pelvic pain, obstructive malformations, and/or other pathologic conditions of the reproductive tract are included
in the differential diagnosis. Nongynecologic causes of pelvic pain (such as, constipation, Crohn disease, and irritable bowel syndrome)
should be considered.
Management
Primary Dysmenorrhea
• Prostaglandin synthetase inhibitors provide relief in 70% to 80% of patients (Laufer, 2012). They should be administered at
onset of menses or, if cramping precedes menses, at onset of symptoms. Treat the patient for the duration of the pain, usually 1
to 2 days. The trial period should extend for three cycles; if no relief is experienced, an alternative prostaglandin inhibitor
should be tried. See Box 36-9 for specific prostaglandin inhibitors. Nonsteroidal anti-inflammatory drugs (NSAIDs) are
advantageous as first-line therapy, because they need to be taken for only 2 to 3 days. Ibuprofen and naproxen are widely used
in clinical practice, but if ineffective, use one of the fenamates. Taking NSAIDs with food helps prevent abdominal complaints.
• OCPs are widely used for dysmenorrhea. Because they suppress ovulation, total progesterone-induced prostaglandin
production is decreased in the endometrium. A 30- to 35-mcg estrogen-progestin combination pill may be used for a 3- to 6-
month trial if 969prostaglandin inhibitors are not successful (Laufer, 2012). The Cochrane Review Group (Wong et al, 2009)
found OCPs may be more effective for dysmenorrhea than placebo; however, interpretation was limited due to the variable
quality of the randomized controlled trials (RCTs) reviewed.
• CAM is likely to be beneficial per the Cochrane Review Group (Proctor and Farquhar, 2004) (see Chapter 43 for further CAM
therapies).
• Application of topical heat
• Thiamine 100 mg/day
• Toki-shakuyaku-san (herbal remedy) 2.5 g three times daily
• High-frequency transcutaneous electrical nerve stimulation (TENS)
• Vitamin E, 500 units/day
• Magnesium
• Follow up by telephone or visit to adjust dose or change medication as needed; the adolescent should be seen again in 3 to 4
months. If failure to respond after 6 months of treatment or if pain worsens over time, a further workup is warranted.Coarctation of the Aorta
Coarctation of the aorta (COA) is a narrowing of a small or long segment of the aorta (Fig. 31-16). Coarctation may occur as a single defect
caused by a disturbance in the development of the aorta or may be secondary to constriction of the ductus arteriosus. The severity of the
coarctation, its location, and the degree of obstruction determine the clinical presentation. Systolic and diastolic hypertension exists in vessels
proximal to the narrowing, whereas hypotension is present in vessels below the narrowing. COA accounts for 6% to 8% of all congenital
heart defects and occurs slightly more in males (Beckman, 2013). The incidence in females with Turner syndrome is close to 30%; it is also
commonly associated with a bicuspid aortic valve in greater than 50% of cases (Park, 2014).
FIGURE 31-16 Coarctation of the aorta (COA). (From Hockenberry M, Wilson D: Nursing care of infants and children, ed 10, St. Louis, 2015, Mosby/Elsevier.)
Clinical Findings
History.
In newborns, COA is not always apparent until the ductus closes and decreases blood flow to the lower body. Severe coarctation in infants is
apparent in the first 6 weeks; symptoms include tachypnea, poor feeding, and possibly cool lower extremities. In children 3 to 5 years old,
coarctation may go unnoticed until hypertension or a murmur is detected. Retrospectively, children with coarctation may have had complaints
of leg pain with exercise or headaches.
Physical Examination.
• Upper extremity hypertension with lower extremity hypotension are present, although milder cases may cause only a minimal discrepancy
between upper and lower extremity BPs. In severe cases, poor lower extremity perfusion may be noticed with lower body mottling or pallor.
• Delayed timing and absent or weak arterial and other distal arterial pulses may occur.
• Bounding brachial, radial, and carotid pulses may occur.
• Signs of CHF may be evident.
• A systolic ejection murmur may be detected in the left infraclavicular region with transmission to the back.
• A ventricular heave at the apex may be palpated.
• A gallop rhythm may occur in infants with CHF.
Diagnostic Studies.
• Chest radiography may reveal a normal or slightly enlarged heart and normal to increased pulmonary vascular markings; rib notching may
be seen.• ECG findings depend on the severity of the lesion and the age of the child. In infants, right ventricular 780hypertrophy may be seen; in older
children, LVH develops secondary to hypertension.
• Echocardiography is helpful in confirming the diagnosis and locating the constricted aortic segment. It may also show associated cardiac
abnormalities. In newborns with a PDA diagnosis by echocardiogram can be challenging.
• MRI can define the location, severity, and anatomy of the aortic arch.
Management
• In critical neonatal coarctation, PGE1 is used to maintain or reopen the ductus.
• If possible, surgical resection of the constricted area and anastomosis of the upper and lower portions of the aorta is performed. Restenosis is
more likely to occur if repair was before 1 year old (Park, 2014). Cardiologists may dilate or stent the coarcted area in recoarctation or mild
coarctation. Other procedures, including bypass grafting, may be necessary with unusually long coarcted segments. Surgical mortality is
rare. Some centers choose balloon valvuloplasty or stent procedures for initial coarctation management (Park, 2014).
• In older children with long-standing hypertension, antihypertensive medication may be required for several months after repair. Long-term
prognosis is excellent unless there are associated intracardiac defects. BP should be monitored postoperatively for recoarctation.
• Children with previous coarctation repairs may participate in any competitive sport if residual BP gradient between arm and legs is less than
20 mm Hg and peak systolic BP is normal at rest and with exercise. However, during the first year after surgery, high-intensity static
exercises, such as weight lifting and wrestling, should be avoided (Park, 2014).
• Lifelong follow-up is necessary due to risk of recoarctation, residual hypertension, and often associated bicuspid aortic valve.
• SBE prophylaxis is no longer considered necessary except in the 6-month postoperative period or if prosthetic material is used (see Box 31-
3).
Aortic Stenosis and Insufficiency
Aortic stenosis or narrowing may occur at the aortic valvular, subvalvular, or supravalvular level. Valvular stenosis is the most
common form (Fig. 31-15). The stenotic aortic valve is usually bicuspid rather than tricuspid. Stenosis causes increased pressure
load on the left ventricle leading to LVH and, ultimately, ventricular failure. The imbalance between increased myocardial
oxygen demand of hypertrophied myocardium and coronary blood supply may lead to ischemia and fatal ventricular
arrhythmias. The bicuspid aortic valve generally becomes more stenotic and often regurgitant (insufficient) over time. Some
infants are born with critical aortic stenosis and require urgent intervention, usually a balloon valvuloplasty early in life.
Children with only a congenital bicuspid aortic valve and no stenosis or 778regurgitation are at risk of developing symptoms by
adolescence. Aortic stenosis occurs in 3% to 8% of all CHDs with a male to female ratio of approximately 4 : 1 (Schneider and
Moore, 2013).Thirty percent of females with Turner syndrome have obstruction at some level of the left heart outflow tract
(Richards and Garg, 2010).
FIGURE 31-15 Aortic stenosis. (From Hockenberry M, Wilson D: Nursing care of infants and children, ed 10, St. Louis, 2015, Mosby/Elsevier.)Clinical Findings
History.
• Growth and development may be normal.
• Activity intolerance, fatigue, chest pain (angina pectoris), or syncope can develop or increase with age.
• CHF, low cardiac output, and shock may be evident in newborns with severe aortic stenosis.
• Sudden death, presumably due to arrhythmias, can occur with increasing severity of stenosis and exertion.
Physical Examination.
• BP may reveal a narrow pulse pressure. The apical impulse may be pronounced with moderate to severe stenosis.
• A grade III to IV/VI, loud, harsh systolic crescendo-decrescendo murmur is best heard at the upper right sternal border with radiation to the
neck, LLSB, and apex.
• With a valvular lesion, a faint, early systolic click at the LLSB may be heard.
• With aortic insufficiency, an early diastolic blowing murmur is heard at the LLSB to apex.
• In the most severe lesions, S2 is single or closely split; S3 or S4 heart sounds may also be heard.
• A thrill may be present at the suprasternal notch.
Diagnostic Studies.
• Chest radiographs are usually normal or may show LVH. Adults frequently develop radiographic evidence of calcification on the aortic
valve over time.
• ECG can be normal or reveal LVH and inverted T-waves.
• A 24-hour Holter monitor or 30-day event monitor demonstrates ventricular arrhythmia.
• Echocardiogram is the diagnostic examination of choice.
Management
• The type and timing of treatment depends on the severity of the obstruction.
• Balloon valvuloplasty of the stenotic valve is the initial palliative treatment in the newborn. However, the aortic valve generally needs
further intervention.
• In older children, surgical division of fused valve commissures may relieve stenosis but often valve replacement is necessary for severe
aortic stenosis and/or insufficiency. Unfortunately, none of the current replacement options are ideal or enduring for children. Mechanical
valves are prothrombotic and require anticoagulation with warfarin. Heterograph and homograft valves have limited durability in the aortic
position, and the Ross procedure requires placement of the homograft in the pulmonic position, leading to future replacements of that valve
as it becomes stenosed.
• Children with subaortic stenosis require surgical resection when the gradient is greater than 35 mm Hg.
• Patients with supravalvar aortic stenosis require resection of the narrowed area with patch material.
• Children with mild aortic stenosis can participate in all sports but should have annual cardiac examinations. Those with moderate aortic
stenosis should choose low-intensity sports (such as, golf, bowling, table tennis, or softball) as guided by their cardiologist. Children with
severe aortic stenosis or moderate aortic stenosis with symptoms should avoid competitive or intensive sports because of the risk of sudden
death from ventricular arrhythmias (Park, 2014) (see Chapter 13, Table 13-6).
• Any aortic root dilation (commonly seen with bicuspid or stenotic aortic valves) may require intervention to prevent aortic dissection.
• SBE prophylaxis is necessary for 6 months after surgery.
• Anticoagulation is necessary with mechanical valve replacement (Park, 2014).
Mitral valve
prolapse
Positive family
history
Midsystolic click; thin; thoracic skeletal
abnormalities
Inverted T waves in
aVF
Normal except skeletal
anomalies
Headaches
Headaches of all types are one of the most common reasons parents seek medical care for their children (Raieli et al, 2010). They
are common during childhood, increasing in frequency and incidence during adolescence. Headaches fall into two
classifications—acute and chronic. Box 28-4 lists the more common types found in these classifications. A person mayexperience different types of headaches, and migraines may be particularly difficult to diagnose because they can be expressed
differently and incompletely during childhood.
The exact physiologic mechanism and etiology for many headaches have not been conclusively determined. Headache pain occurs when
pain-sensitive intracranial structures are activated. Such structures include the arteries of the circle of Willis and some of their branches,
meningeal arteries, large veins and dural venous sinuses, and part of the dura near blood vessels. Muscles around the head, neck, scalp, eyes,
jaw, teeth, sinuses, and the external carotid artery and its branches are pain sensitive structures external to the skull. Stimulation of these
structures results in more localized pain that is carried by CN V, CN VII, CN IX, and CN X. In contrast, intracranial stimulation refers pain
imprecisely (e.g., occipital lobe tumor).
Studies indicate that 40% of children will experience a headache by 7 years old and 75% by 15 years old (Rubin et al, 2010). Prevalence
rates for migraine headaches are reported to be: age 3 (3% to 8%), age 5 (19.5%), age 7 (37% to 51%), and 7 to 15 years old (57% to 82%).
Before 10 years old, the incidence is higher in males than females. During teenage years, females have a higher headache incidence. The
mean age at onset of migraine is 7.2 years old for males and 10.9 years old for females (Lewis et al, 2004).
The provider must discern between symptoms that suggest that a headache is primary (e.g., tension-type, cluster, migraine type) or due to
a secondary cause (e.g., tumor, hydrocephaly, infection, intoxication [lead, carbon monoxide], idiopathic intracranial hypertension, increased
intracranial pressure). Key historical questions and a thorough workup are mandatory in order to exclude secondary headache etiology. In the
absence of findings suggestive of a secondary headache, a more certain diagnosis of a primary headache disorder can be made. The
International Headache Society (IHS) provides succinct clinical criteria (available at www.ihs-classification.org/en/) to help the provider
evaluate, delineate between, and classify primary headaches (e.g., including migraines with or without aura and migraine subtypes) and
secondary headaches.
Clinical Findings
History.
Headache diaries may be used to gather history and track symptoms over time. Some useful ones are available for downloading
at www.achenet.org/resources/headache_diaries/. Important questions to ask the child and parent(s) include:
• Duration: Recent severe onset is worrisome.
• Frequency and triggers: Children with recurrent, low-intensity headaches, with no neurologic changes, and who recover completely between
episodes are unlikely to have serious intracranial etiology. Triggers can include ovulation or menstruation, exercise, food or odors, and
stress. Other triggers can include chocolate, processed meats, aged cheeses, nuts, altered amounts of caffeine intake, dairy products,
shellfish, and some dried fruits. Consistent findings such as perimenstrual exacerbation, food triggers, and a stable pattern to the headache
with intervals of wellness over a long time period are reassuring symptoms that suggest a primary headache. 686In most cases, a specific
trigger or etiology is not ever identified.
• Location: Occipital or consistently localized headaches can indicate underlying pathology. Facial pain might be sinusitis. Ocular motor
imbalance can produce a dull periorbital discomfort, whereas temporomandibular joint pain tends to localize around the periauricular or
temporal areas.
• Quality and severity of pain: Sharp, throbbing, or pounding pain is vascular (migraine). Dull and constant pain may be tension or organic.
Severity can be assessed by asking about limitations to activities and missed school days, although there are other factors that contribute to
missed school and limited activities. How many ―different kinds of headaches‖ are experienced?
• Age of onset: Progression of the headaches over time and longest period of time without symptoms.
• Home management and medication dosages, including dosage and self-management activities.
• Associated symptoms can include nausea, vomiting, visual changes, dizziness, paresthesia, neck/shoulder pain, back pain, otalgia,
abdominal pain, hypersomnia, food cravings, confusion, ataxia, pallor, photophobia, and phonophobia. Changes in gait, personality, vision,
mentation, or behavior that do not occur at the same time as the headache are worrisome and merit further evaluation with referral. There
are some precursor symptoms and conditions that can indicate a predisposition to migraines. These include cyclic vomiting (see Chapter
33), abdominal migraine (see Chapter 33), and BPV. Alone, they do not warrant extensive or expensive workups unless the diagnosis is
unclear. These conditions may evolve into migraine without aura in later childhood (Hershey, 2011; Lewis et al, 2008a).
• Head trauma: If associated with headache, a subdural hematoma or postconcussive syndrome must be considered.
• Psychologic symptoms: Evaluate for the presence of depression, school stressors, or concerns about family functioning. Additional things to
consider include bullying or peer issues at school, ―over programming‖ and family expectations, and meal, hydration, and sleep status.
• Family history: Some children with headache, especially migraine, have a family history of headaches.
Distinguishing Features of Headache Types
• Migraine and migraine with aura: These can be differentiated by the presence or absence of aura symptoms (Table 28-6).
Characteristics of migraines include nausea, abdominal pain, vomiting, unilateral pain, pulsating pain, relief with sleep, an
aura, visual changes such as dark or blind spots, and a history of a family member (usually on the maternal side) with
migraine without aura. Dizziness and motion sickness may be described. Infants and toddlers may present with irritability,
sleepiness, and pallor. In preadolescents, common migraine symptoms are more likely. Nausea and vomiting might not occur,
and the pain can be more frontal. Lethargy and sleep can follow. Visual changes are rare, and the pain quality is variable.
Times between headaches are pain free.• Abdominal migraine: This is rare and is a somewhat controversial diagnosis; symptoms include midline pain, nausea, and
vomiting with minimal or no headache. 687Such symptoms can also be suggestive of complex partial seizures.
• Muscle contraction or tension headaches: The pain is dull and bifrontal or occipital, with nausea and vomiting occurring only
rarely; there is no prodrome. Tension headaches can last for days or weeks but generally do not interfere with activities. In
children, it can be difficult to differentiate migraine and tension-type headaches. Psychosocial stress seems to be a major factor
in tension and chronic daily headaches in both children and adolescents.
• Secondary headaches (or those headaches that have a pathologic process): Key historical markers are sudden onset of
hyperacute or increasing pain severity or accompanying neurologic signs. These require prompt referral. Box 28-5 presents red
flag warnings of a pathologic process indicating immediate referral. Presenting symptoms of these headaches include the
following (Lewis et al, 2008b; Sprague-McRae et al, 2009):
• Headache pain that is worse in the morning on awakening and standing up, and then fades; increases in frequency and
severity over a period of only a few weeks; persistent and unilateral
• Pain that wakens the child from sleep
• Vomiting but not nausea; vomiting may relieve the headache
• Visual disturbances, diplopia, edema of the optic disc (papilledema)
• Increased pain with straining, sneezing, coughing, defecation, or changes in position
• Occipital region and neck pain
• Educational, mental, personality, or behavioral alterations; irritability
• Seizures
• Unsteadiness or dramatic changes in balance
• Fever
• Family history of neurologic disorders (e.g., brain tumors, neurofibromatosis, vascular malformations)
• Child has a history of a ventriculoperitoneal shunt, meningitis, hydrocephaly, or tumor
Physical Examination.
A complete physical and neurologic examination is in order:
• Blood pressure, supine and standing with 2-minute interval between them
• Height and weight
• Head circumference (all children)
• Eyes: Palpate for tenderness; check discs for papilledema, movements
• Ears: Patency of canals, normal tympanic membranes
• Neck: Palpate muscles; check range of motion for nuchal rigidity
• Sinuses (frontal and maxillary)
• Teeth (percuss, inspect)
• Temporomandibular joints (mouth and jaw): Palpate and check range of motion
• Thyroid gland
• Bones and muscles of skull: Palpate for tenderness; listen for cranial bruits; check range of motion of cervical spine
• Extremities: Tandem gait
• Nerves: Palpate supraorbital, trochlear, occipital nerves; assess CN IX to CN XII
• Reflexes: Pronator drift test (Romberg)
• Vision screen
Diagnostic Studies.
Imaging studies are rarely indicated unless the history suggests intracranial pressure (see Box 28-5); there is a sudden onset,
increased severity, or change in headache pattern; the neurologic examination is abnormal; or when a complaint of “dizziness”
fits the criteria listed in Table 28-7. CTs are generally out of favor due to radiation. MRI is the first-line treatment unless
extremely urgent and it cannot be obtained immediately. If abnormal, an MRI should be done. An EEG should be obtained if the
history and physical examination suggest a seizure process. 688If there is a history of external trauma, such as from a motor
vehicle accident, cervical and spinal x-rays should be ordered.
Differential Diagnosis
The differential diagnosis consists of sinusitis, trigeminal neuralgia, pseudotumor cerebri, sleep disorder, hyperthyroidism,
hypertension, cyclic vomiting, abdominal migraine, BPV, and temporomandibular joint dysfunction. Brain tumors, abscesses,hematomas, and arteriovenous malformations in children are generally associated with ataxia, papilledema, intellectual
changes, or behavioral changes. These processes are termed space occupying lesions because they crowd out other intracranial
structures, precipitating edema and interfering with the normal actions of CSF and vessels. Infants may initially accommodate
well to the increase in intracranial pressure because of the ability of their cranial sutures to expand. Visual acuity is rarely a
cause of headaches. Determining the correct headache classification or entity is also part of the differential diagnosis. These and
other causes of headaches in children are outlined in (Table 28-8).
Management
For nonorganic headaches, there may be no known etiology (e.g., no tumor, aneurysm, or metabolic or structural cause). The
child and parents should be taught pain and stress management techniques; nonsteroidal anti-inflammatory drugs are the firstline pharmaceutical for acute treatment. There can be significant loss of school attendance as a result of headaches, but
attendance should be mandatory. A quiet 689rest period may be allowed at school if needed, and school nurses can be helpful in
developing a plan for this. If the child remains home, activities should be restricted to bed and all homework completed. The
child should be returned to school if the pain improves during the school day. Minimize attention to the headache. Relaxation
exercises or biofeedback training can be helpful. Trigger factors should be avoided, if possible.
The goals of treating acute-onset migraines include:
• Abortive therapy
• Reducing frequency, severity, and length of treatment
• Reducing impairment
• Improving overall quality of life
• Avoiding escalation of medications
• Optimizing self-care abilities of the patient and family
• Using beneficial and cost-effective treatment
• Minimizing medication side effects
Many of the newer medications for migraines (e.g., triptans) have not been adequately tested for safety and efficacy in
children and adolescents, with the exception of sumatriptan and zolmitriptan (refer to Table 28-9 for treatment options). Lewis
(2009) recommends treating throughout the school year and then gradually curtailing daily agents during the summer months.
An alternative for younger children is to use shorter courses of preventive medications (6 to 8 weeks) followed by gradual
weaning. All individuals with migraines benefit from regular sleep, exercise, moderate caffeine intake, and adequate hydration.
Medications should be taken as soon as possible after the onset of the headache; should be taken in the prescribed dosage;
should be available at home, school, or work; and the overuse of analgesics is to be avoided (more than three doses per week).
Prophylactic therapy is considered when migraines cause a child to miss school regularly and when the child suffers severe migraine
headaches two to four times a month or tension or migraine three to four times per week with a clear sense of functional disability. The aim
of prophylactic treatment is to reduce headache severity, frequency, or both (El-Chammas et al, 2013). Medication classifications to consider
include beta-blockers, antidepressants, anticonvulsants that treat headaches also, or calcium channel blockers.
The use of magnesium oxide, CoQ10, and riboflavin as dietary supplements is gaining popularity in practice due to tolerance, cost, and
ease of use. However, the evidence in favor of these modalities is limited, and further research is warranted (Orr and Venkateswaran, 2014).
Refer all patients with organic (structural) headaches. Parents seek medical attention for pain relief for their child, in addition to
reassurance that there are no intracranial processes occurring (brain tumors). Each child with headaches requires an individually tailored
strategy that may include pharmacologic and nonpharmacologic modalities.
Complications
School absence and depression are known complications.
[Show More]
![Preview of NR 602 MIDTERM EXAMS STUDY GUIDE [Recently Updated]](https://scholarfriends.com/storage/NR 602 Midterm study guide.png)














![Preview of WEEK **5 [Chamberlain university]NR 602 IHUMAN Case Week #5 Deborah Arnaudin 54 Year Old](https://browseimages.nyc3.digitaloceanspaces.com/paper-images/2026/03/31/zbQKePwt2026-03-31-11-4669cc328fcad96.png)